


A síndrome de DiGeorge (SDG) é uma condição causada por uma microdeleção na localização q11.2 do cromossoma 22 (por isso, também chamada de síndrome de deleção 22q11.2). Há uma alteração do desenvolvimento da terceira e quarta bolsas faríngeas, levando à hipoplasia do timo e da paratiroide (causando imunodeficiência de células T e hipocalcemia, respetivamente). As alterações conotruncais, que se apresentam como defeitos cardíacos congénitos, também são características da doença. Outras manifestações consistem em características faciais típicas, infeções frequentes e distúrbios neuropsiquiátricos. O diagnóstico é obtido por uma combinação de achados clínicos, exames laboratoriais (redução da contagem de células T e cálcio baixo), ecocardiograma e análise genética. O tratamento pode incluir suplementação de cálcio, antibióticos profiláticos, cirurgia (para defeitos cardíacos e alterações do palato) e transplante de timo ou de células hematopoiéticas. O prognóstico depende da gravidade da alteração cardíaca e da imunodeficiência.
Last updated: Dec 15, 2025
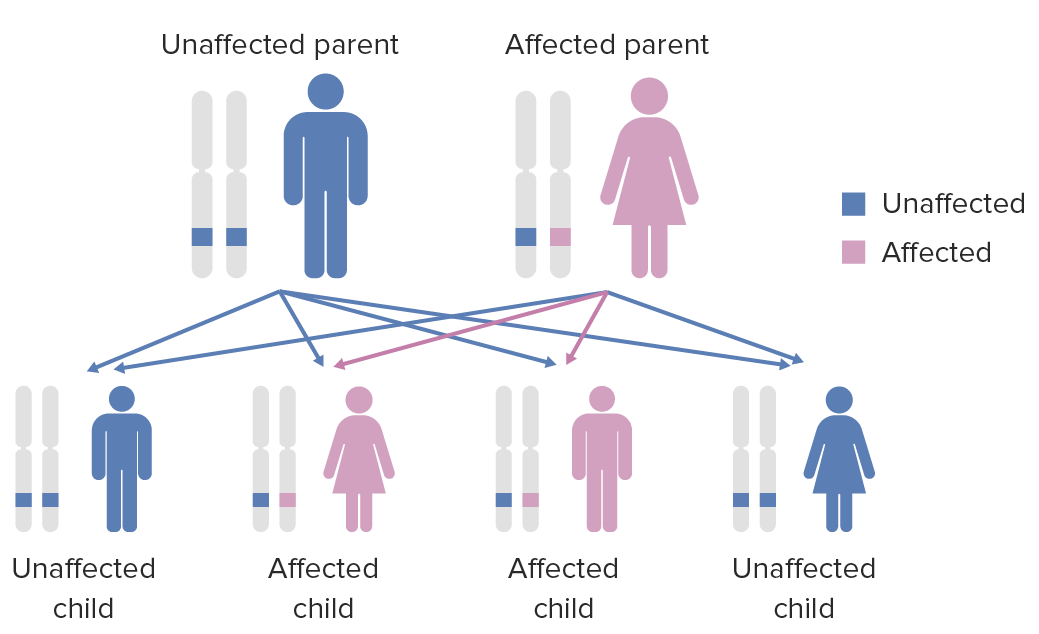
Diagrama do padrão de hereditariedade de condições autossómicas dominantes
Imagem por Lecturio.
Síndrome de DiGeorge: deleção em 11.2 do braço q
Imagem por Lecturio.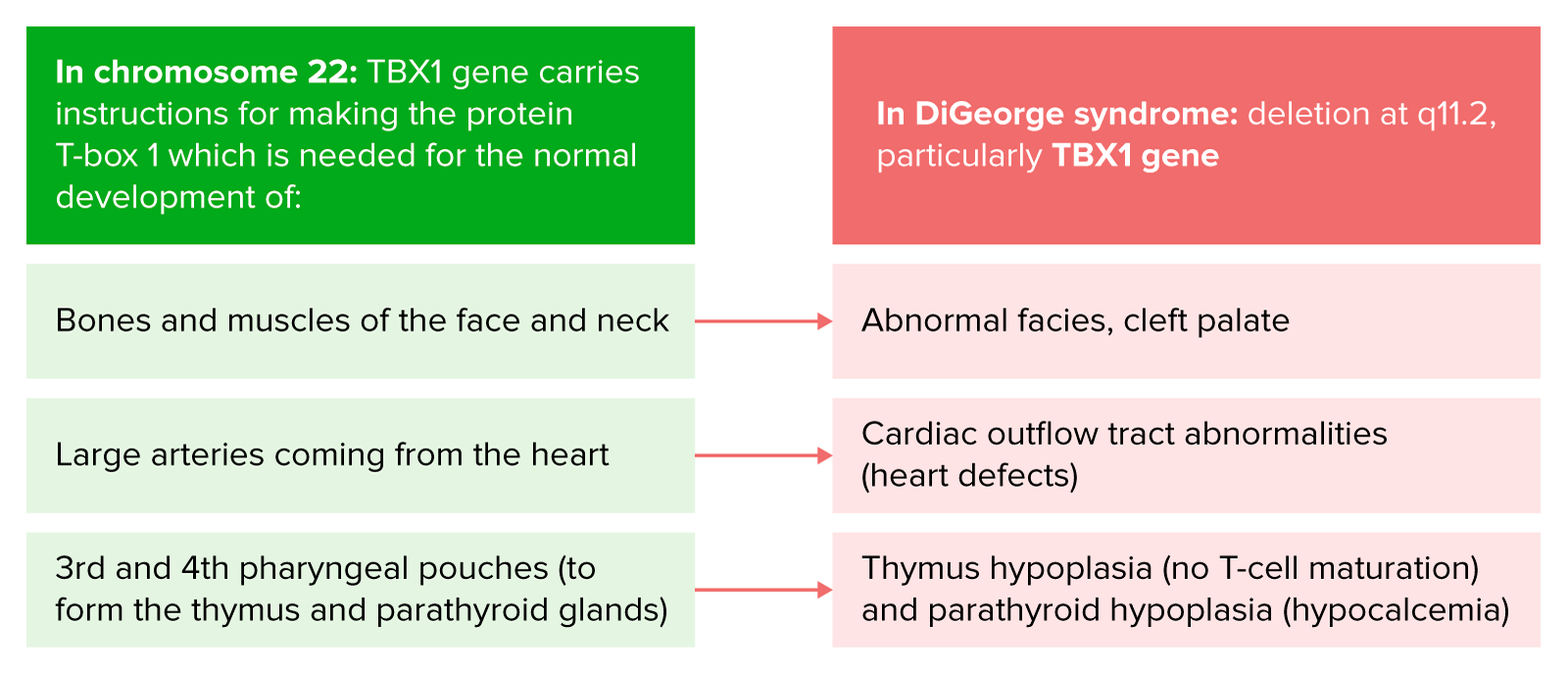
Fisiopatologia da síndrome de DiGeorge
Imagem por Lecturio.A síndrome de DiGeorge tem uma variabilidade acentuada na expressão clínica entre diferentes indivíduos. As manifestações podem incluir o seguinte:
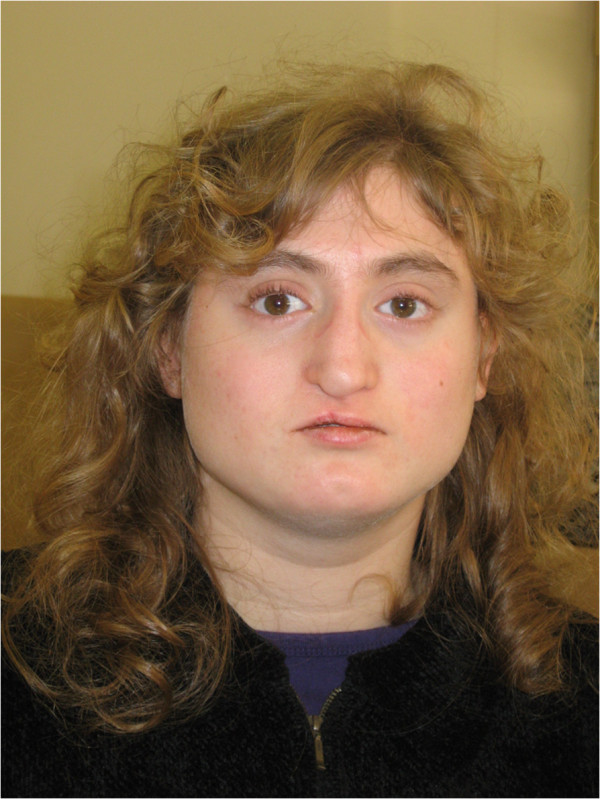
Aparência facial de um doente com deleção 22q11.2 atípica. Observar o rosto largo e quadrado; hipotelorismo; base nasal estreita e ponte nasal larga; ponta nasal saliente; ponta nasal desviada (estado pós-reparação de lábio leporino); filtro curto; boca estreita; covinhas no queixo; e pescoço largo.
Imagem: “Facial appearance of Patient VC901” por Elena Michaelovsky et al. Licença: CC BY 2.0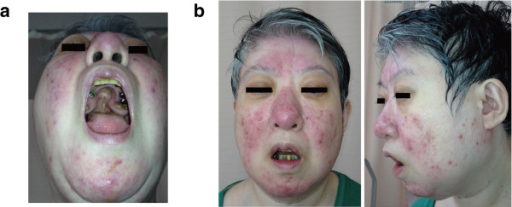
Características da deleção 22q11.2 numa mulher de 48 anos.
(a) fenda palatina; realizou intervenção cirurgia anterior
(b) Características faciais dismórficas leves, incluindo linha anterior do cabelo baixa, pálpebras edematosas, achatamento malar, nariz com ponta nasal bulbosa, asa nasal hipoplásica e raiz nasal quadrada e achatada, boca pequena e lábio superior fino
Os sinais da síndrome de DiGeorge podem ser resumidos usando a mnemónica CATCH-22:
Estabelecer um diagnóstico é difícil devido à variabilidade dos fenótipos. O diagnóstico da síndrome de DiGeorge é determinado pela demonstração de uma diminuição das células T CD3 CD3 Complex of at least five membrane-bound polypeptides in mature T-lymphocytes that are non-covalently associated with one another and with the T-cell receptor. The CD3 complex includes the gamma, delta, epsilon, zeta, and eta chains (subunits). When antigen binds to the T-cell receptor, the CD3 complex transduces the activating signals to the cytoplasm of the T-cell. The CD3 gamma and delta chains (subunits) are separate from and not related to the gamma/delta chains of the T-cell receptor. T cells: Types and Functions+, para além dos achados clínicos característicos e estudos genéticos que demonstram a deleção na área cromossómica 22q11.2.
O tratamento visa tratar as características associadas à doença. A intervenção precoce e a avaliação do desenvolvimento são fundamentais.
Não há cura para a síndrome de DiGeorge. A expectativa de vida depende em grande parte da gravidade dos defeitos cardíacos (fator mais MAIS Androgen Insensitivity Syndrome importante) e da imunodeficiência.
