CHARGE syndrome is a rare genetic condition with autosomal dominant inheritance Autosomal dominant inheritance Autosomal Recessive and Autosomal Dominant Inheritance in which almost all body systems are affected. The acronym CHARGE stands for the constellation of clinical features seen with this condition: Coloboma, Heart defects, Atresia choanae, Growth retardation, Genital abnormalities, and E ar AR Aortic regurgitation (AR) is a cardiac condition characterized by the backflow of blood from the aorta to the left ventricle during diastole. Aortic regurgitation is associated with an abnormal aortic valve and/or aortic root stemming from multiple causes, commonly rheumatic heart disease as well as congenital and degenerative valvular disorders. Aortic Regurgitation abnormalities. Genetic testing Genetic Testing Detection of a mutation; genotype; karyotype; or specific alleles associated with genetic traits, heritable diseases, or predisposition to a disease, or that may lead to the disease in descendants. It includes prenatal genetic testing. Myotonic Dystrophies confirms the diagnosis. Treatment is symptomatic with management of the airway Airway ABCDE Assessment, heart defects, and feeding ability as the priorities in early life. There is no definitive curative therapy.
Last updated: Dec 15, 2025
CHARGE is an acronym to describe the clinical presentation of this syndrome:
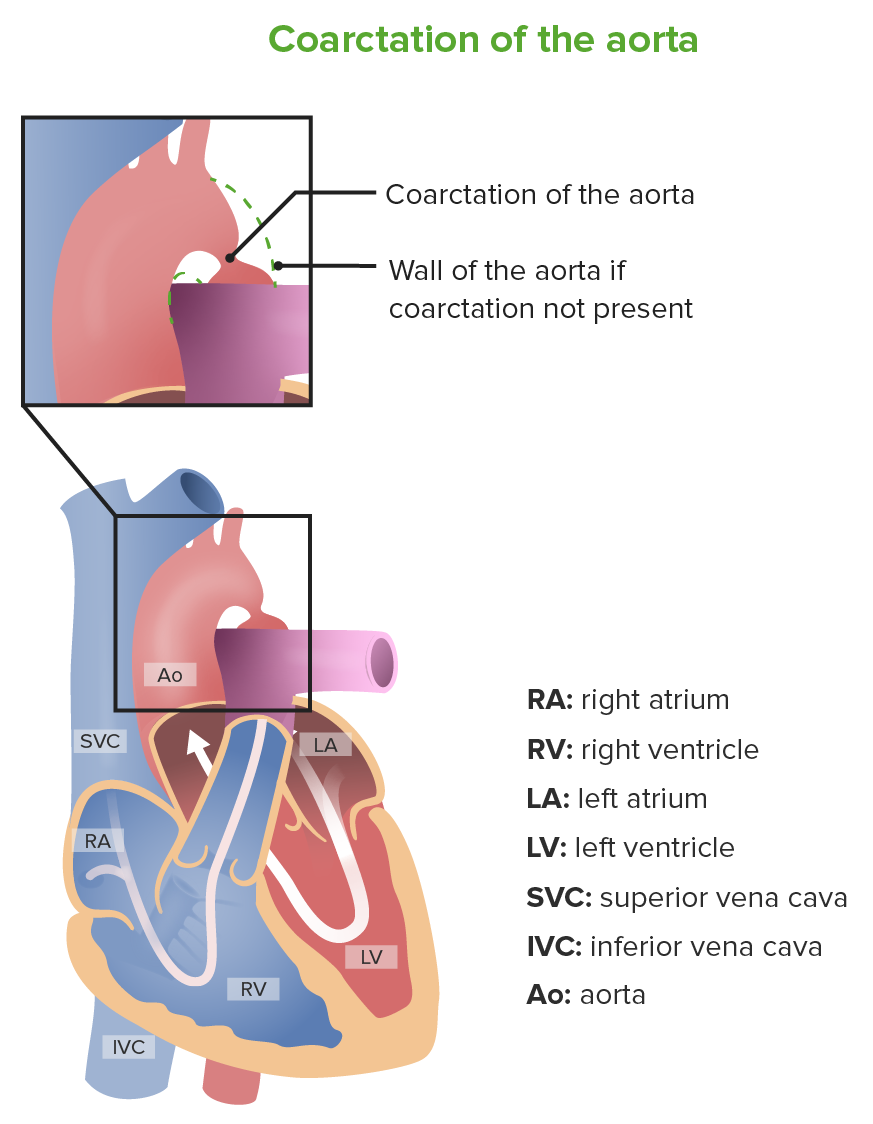
Coarctation of the aorta
Image by Lecturio.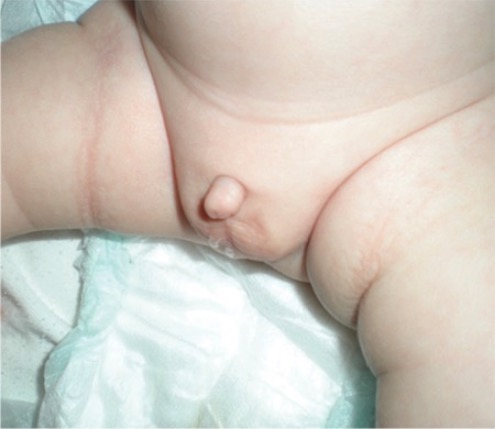
Infant born with a micropenis
Image: “Micropenis in a newborn” by Erciyes University Faculty of Medicine, Department of Pediatric Endocrinology, Kayseri, Turkey. License: CC BY 2.5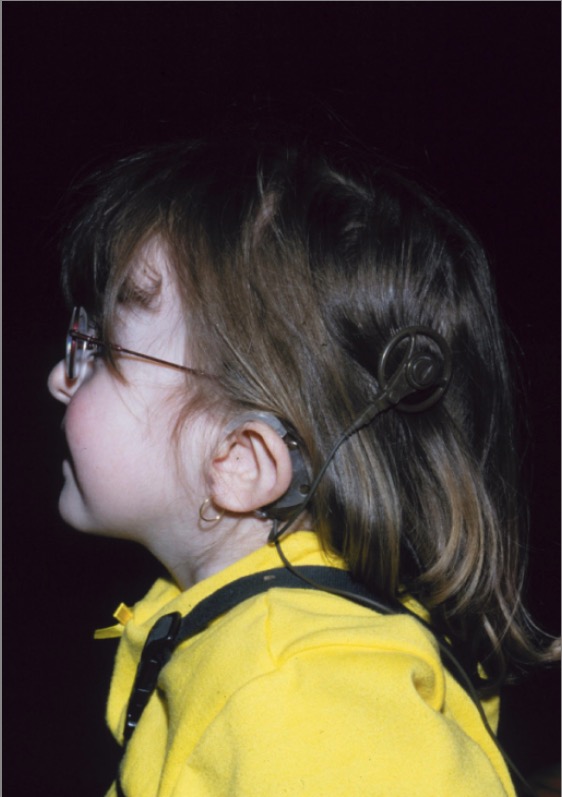
A girl with CHARGE syndrome: Note the so-called “lop ear” characteristic of CHARGE syndrome, along with the cochlear implant.
Image: “CHARGE syndrome” by Department of Pediatrics, IWK Health Centre, Dalhousie University, Canada. License: CC BY 2.0The diagnosis of CHARGE syndrome can be established if 2 major criteria plus any number of minor criteria are present.
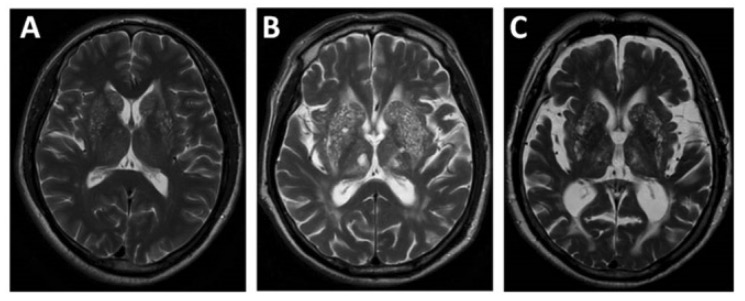
Magnetic resonance imaging showing cerebral atrophy from mild (A) to moderate (B) to severe (C) stages
Image: “EPVS” by Department of Neurology, Beijing Chaoyang Hospital, Capital Medical University, Beijing, China. License: CC BY 4.0Various supportive and corrective treatment options are available. Treatment is tailored to the symptoms and anomalies present.