Esophageal atresia Atresia Hypoplastic Left Heart Syndrome (HLHS) is a congenital anomaly in which the upper esophagus Esophagus The esophagus is a muscular tube-shaped organ of around 25 centimeters in length that connects the pharynx to the stomach. The organ extends from approximately the 6th cervical vertebra to the 11th thoracic vertebra and can be divided grossly into 3 parts: the cervical part, the thoracic part, and the abdominal part. Esophagus: Anatomy is separated from the lower esophagus Esophagus The esophagus is a muscular tube-shaped organ of around 25 centimeters in length that connects the pharynx to the stomach. The organ extends from approximately the 6th cervical vertebra to the 11th thoracic vertebra and can be divided grossly into 3 parts: the cervical part, the thoracic part, and the abdominal part. Esophagus: Anatomy and ends in a blind pouch. The condition may be isolated or associated with tracheoesophageal fistula Fistula Abnormal communication most commonly seen between two internal organs, or between an internal organ and the surface of the body. Anal Fistula, which is an abnormal connection between the trachea Trachea The trachea is a tubular structure that forms part of the lower respiratory tract. The trachea is continuous superiorly with the larynx and inferiorly becomes the bronchial tree within the lungs. The trachea consists of a support frame of semicircular, or C-shaped, rings made out of hyaline cartilage and reinforced by collagenous connective tissue. Trachea: Anatomy and esophagus Esophagus The esophagus is a muscular tube-shaped organ of around 25 centimeters in length that connects the pharynx to the stomach. The organ extends from approximately the 6th cervical vertebra to the 11th thoracic vertebra and can be divided grossly into 3 parts: the cervical part, the thoracic part, and the abdominal part. Esophagus: Anatomy. The most common congenital tracheoesophageal abnormality is a combination of esophageal atresia Atresia Hypoplastic Left Heart Syndrome (HLHS) and distal tracheoesophageal fistula Fistula Abnormal communication most commonly seen between two internal organs, or between an internal organ and the surface of the body. Anal Fistula. The defect manifests early after birth as an inability to feed, regurgitation Regurgitation Gastroesophageal Reflux Disease (GERD), choking, and aspiration, and requires surgical correction. Survival is good, but long-term complications are common.
Last updated: Jan 9, 2025
Tracheoesophageal fistula Fistula Abnormal communication most commonly seen between two internal organs, or between an internal organ and the surface of the body. Anal Fistula (TEF) and esophageal atresia Atresia Hypoplastic Left Heart Syndrome (HLHS) (EA) are congenital malformations Congenital malformations Congenital malformations or teratogenic birth defects are developmental disorders that arise before birth during the embryonic or fetal period. The rate of incidence for children born alive is approximately 3%. Teratogenic Birth Defects of the esophagus Esophagus The esophagus is a muscular tube-shaped organ of around 25 centimeters in length that connects the pharynx to the stomach. The organ extends from approximately the 6th cervical vertebra to the 11th thoracic vertebra and can be divided grossly into 3 parts: the cervical part, the thoracic part, and the abdominal part. Esophagus: Anatomy and lower respiratory tract.
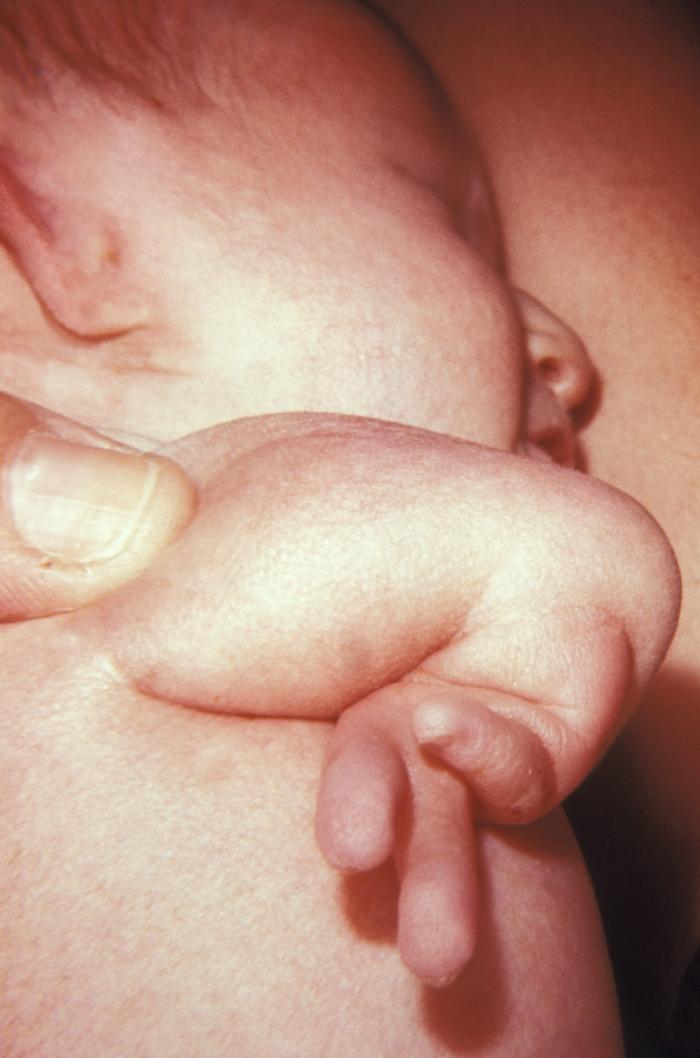
A newborn with radial atresia of the right arm: This birth deformation is a VACTERL association disorder.
Image: “Radial atresia” by CDC/ Dr. James W. Hanson. License: Public Domain| Types | Description | Incidence Incidence The number of new cases of a given disease during a given period in a specified population. It also is used for the rate at which new events occur in a defined population. It is differentiated from prevalence, which refers to all cases in the population at a given time. Measures of Disease Frequency |
|---|---|---|
| Type A | EA without TEF | Approximately 8% of cases |
| Type B | EA with TEF to the proximal esophageal segment | Approximately 1% of cases |
| Type C | EA with TEF to the distal esophageal segment | Approximately 84% of cases |
| Type D | EA with TEF to the proximal and distal esophageal segments | Approximately 3% of cases |
| Type E | H-type TEF without atresia Atresia Hypoplastic Left Heart Syndrome (HLHS) | Approximately 4% of cases |
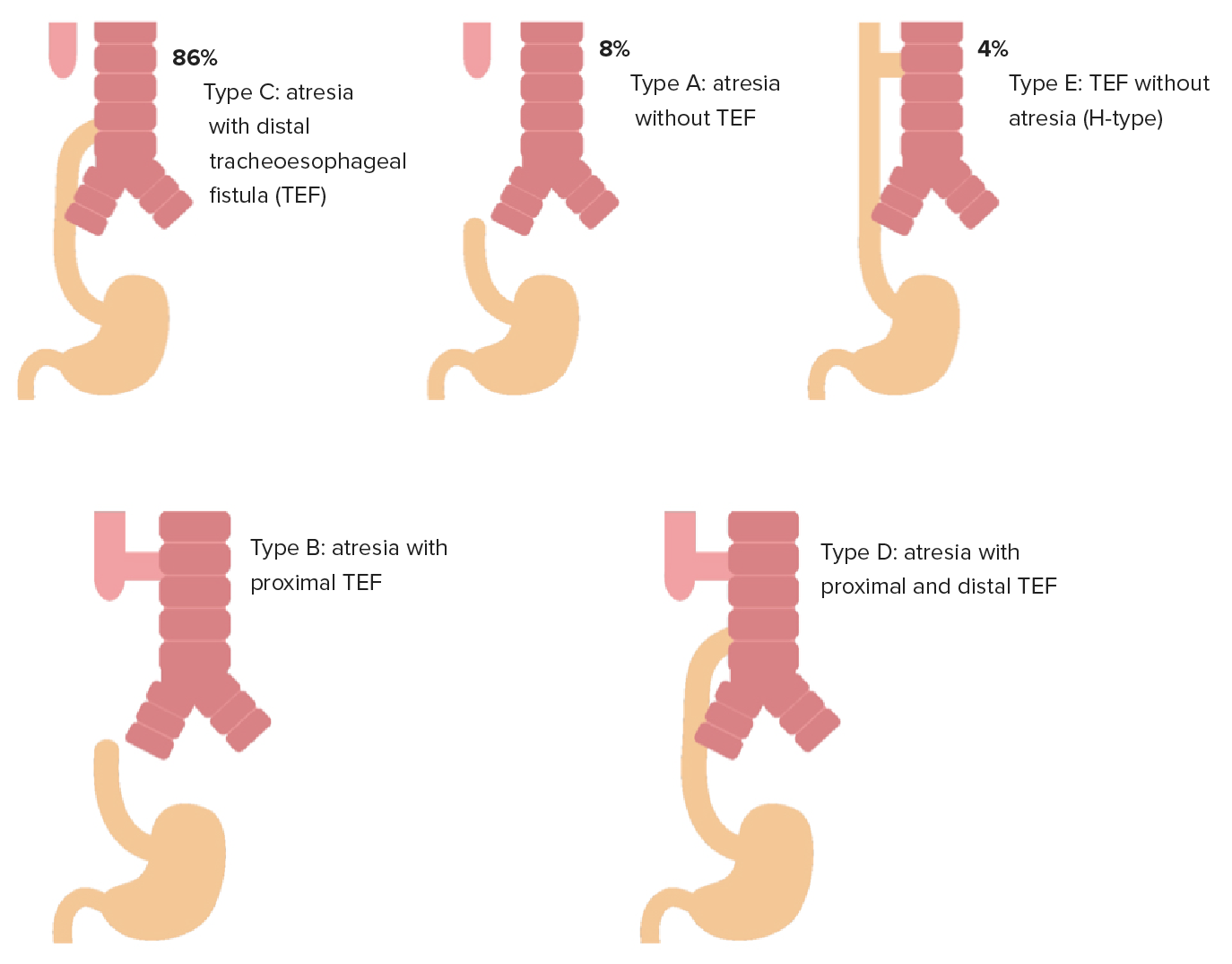
Types of EA with TEF (gross classification): From top left, images in order of incidence: type C (84%), type A (8%), type E (4%); bottom row: type D and type B (least common).
Image by Lecturio.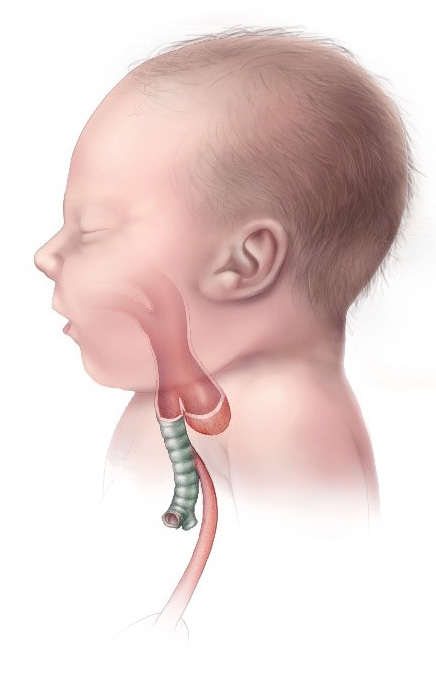
Esophageal atresia and TEF type C (most common type): EA is accompanied by a TEF to the distal esophageal segment.
Image: “Esophageal Atresia TYPE C” by CDC. License: Public Domain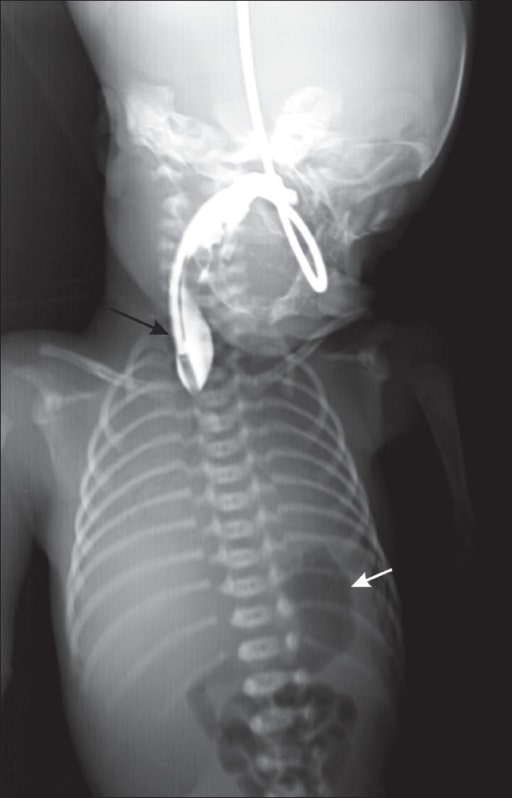
Chest radiograph of a neonate with contrast shows a blind-ending upper end of the esophagus (black arrow) and a gas-filled stomach (white arrow).
Image: “Chest radiograph” by Department of Radiology, Geetanjali Medical College and Hospital, Udaipur, Rajasthan, India. License: CC BY 2.0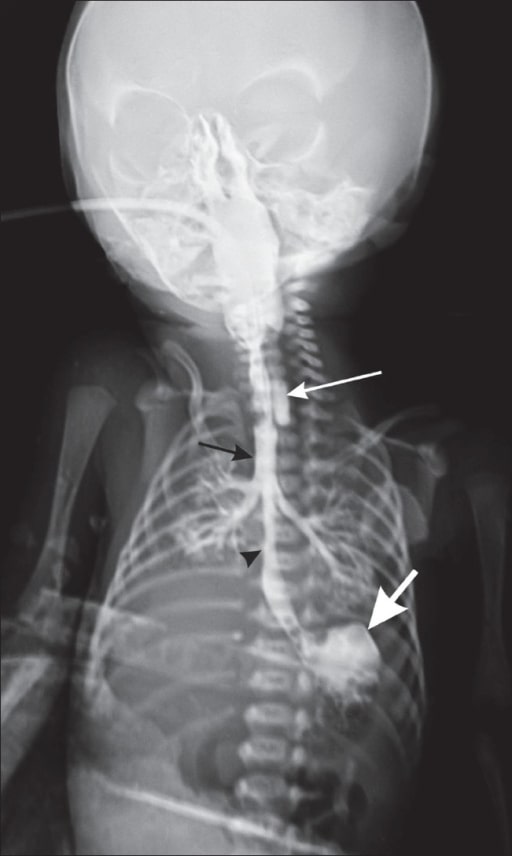
Chest radiograph of a neonate with contrast shows the blind-ending upper end of the esophagus (long white arrow), contrast in the trachea (black arrow), the lower end of the esophagus communicating with the trachea (black arrowhead), and contrast in the stomach (thick white arrow).
Image: “Chest radiograph” by Department of Radiology, Geetanjali Medical College and Hospital, Udaipur, Rajasthan, India. License: CC BY 2.0Supportive:
Surgery (definitive):