


A síndrome de CHARGE é uma doença genética, rara, com hereditariedade autossómica dominante, na qual quase todos os sistemas do corpo são afetados. O acrónimo CHARGE representa a constelação de características clínicas observadas nesta condição: Coloboma, Heart defects, Atresia choanae, Growth retardation, Genital Abnormalities, and Ear abnormalities ( coloboma Coloboma Congenital anomaly in which some of the structures of the eye are absent due to incomplete fusion of the fetal intraocular fissure during gestation. Esophageal Atresia and Tracheoesophageal Fistula, defeitos cardíacos, atrésia das coanas , atraso do crescimento, alterações genitais e alterações do ouvido). O teste genético confirma o diagnóstico. O tratamento é sintomático, sendo a abordagem da via aérea, defeitos cardíacos e capacidade de alimentação as prioridades no período de início de vida. Não existe terapêutica curativa definitiva.
Last updated: Dec 15, 2025
CHARGE é um acrónimo para descrever a apresentação clínica desta síndrome:
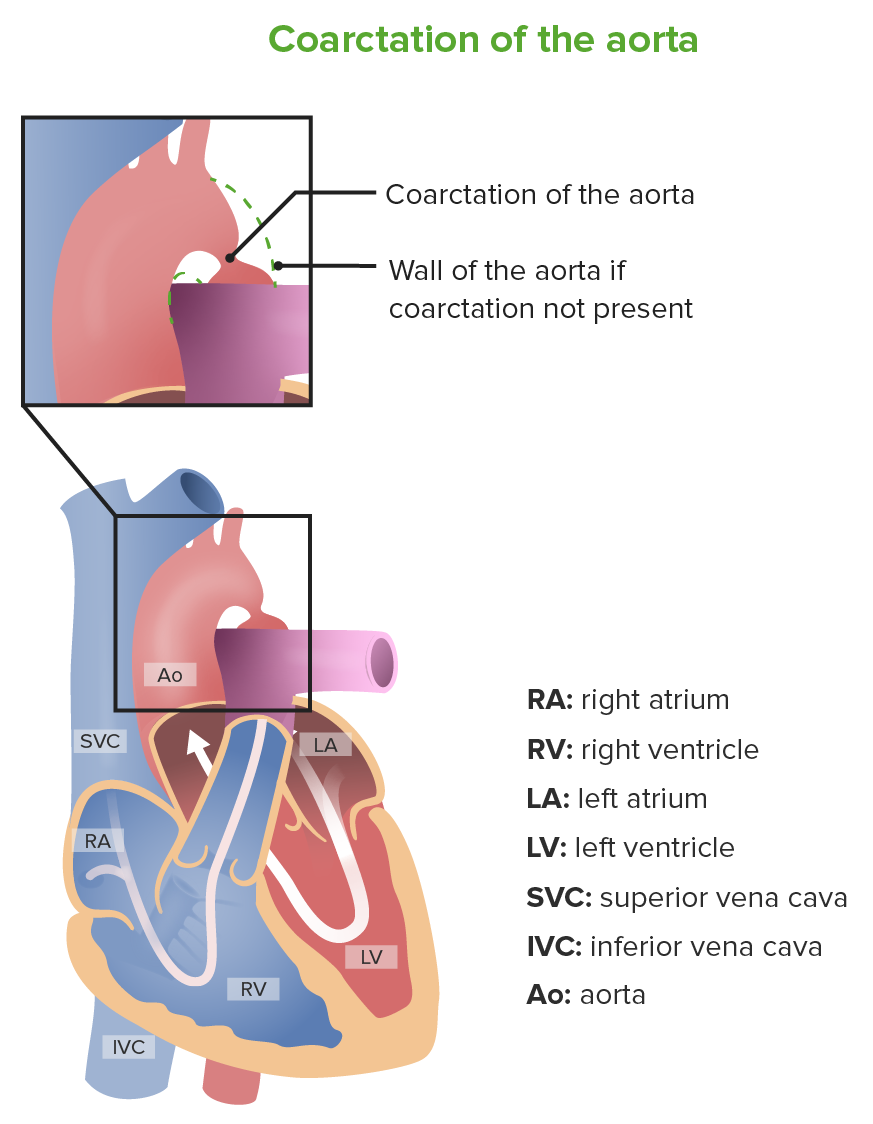
Coartação da aorta
Imagem por Lecturio.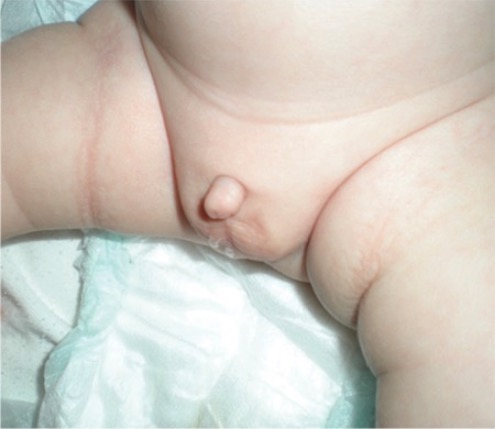
Criança nascida com micropénis
Imagem: “Micropenis in a newborn” por Erciyes University Faculty of Medicine, Department of Pediatric Endocrinology, Kayseri, Turkey. Licença: CC BY 2.5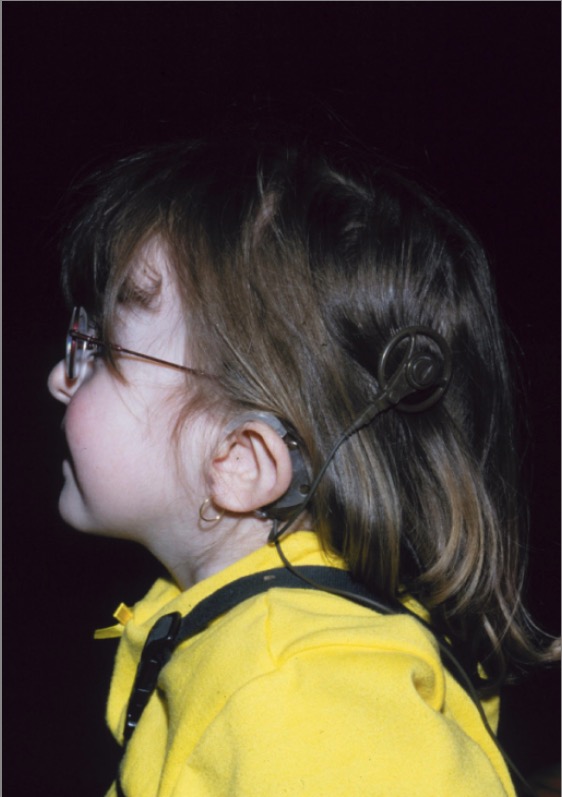
Uma menina com síndrome de CHARGE: Observar a denominada “orelha caída” característica da síndrome de CHARGE, juntamente com o implante coclear.
Imagem: “CHARGE syndrome” por Department of Pediatrics, IWK Health Centre, Dalhousie University, Canada. Licença: CC BY 2.0O diagnóstico da síndrome de CHARGE pode ser estabelecido se 2 critérios major e qualquer número de critérios minor estiverem presentes.
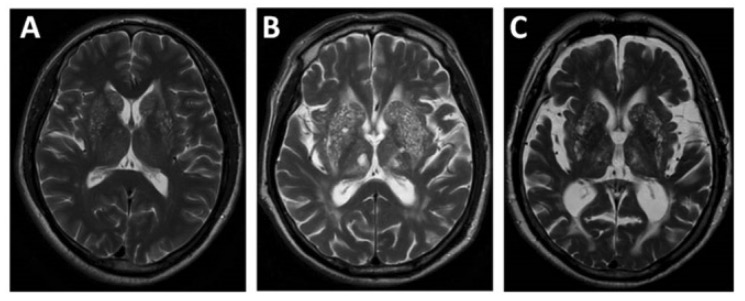
Ressonância magnética a demonstrar atrofia cerebral de estadio leve (A) a moderado (B) a grave (C)
Imagem: “EPVS” por Department of Neurology, Beijing Chaoyang Hospital, Capital Medical University, Beijing, China. Licença: CC BY 4.0Estão disponíveis várias opções de tratamento de suporte e corretivo. O tratamento é adaptado aos sintomas e alterações presentes.
