Astrocytomas are neuroepithelial tumors that arise from astrocytes, which are star-shaped glial cells. Under WHO CNS 5, astrocytomas are classified as pilocytic astrocytomas (grade 1), diffuse astrocytomas (grade 2), anaplastic astrocytomas (grade 3), and glioblastomas (grade 4). Grade 1 tumors are benign and occur in children, whereas IDH‑wildtype glioblastoma is the most common primary malignant brain tumor in adults and carries a poor prognosis. Clinical presentation varies by location and grade but often includes headache, seizures, and focal neurologic deficits. Diagnosis requires MRI combined with histopathologic and molecular analysis of biopsy tissue, and treatment depends on the tumor’s grade and molecular subtype, including surgery, radiotherapy, and chemotherapy.
Astrocytomas are neuroepithelial tumors in the CNS arising from astrocytesAstrocytesA class of large neuroglial (macroglial) cells in the central nervous system – the largest and most numerous neuroglial cells in the brain and spinal cord. Astrocytes (from ‘star’ cells) are irregularly shaped with many long processes, including those with ‘end feet’ which form the glial (limiting) membrane and directly and indirectly contribute to the blood-brain barrier. They regulate the extracellular ionic and chemical environment, and ‘reactive astrocytes’ (along with microglia) respond to injury.Nervous System: Histology, a type of star-shaped glial cell.
Glial cellsGlial CellsThe non-neuronal cells of the nervous system. They not only provide physical support, but also respond to injury, regulate the ionic and chemical composition of the extracellular milieu, participate in the blood-brain barrier and blood-retinal barrier, form the myelin insulation of nervous pathways, guide neuronal migration during development, and exchange metabolites with neurons. Neuroglia have high-affinity transmitter uptake systems, voltage-dependent and transmitter-gated ion channels, and can release transmitters, but their role in signaling (as in many other functions) is unclear.Nervous System: Histology are supportive tissues within the brainBrainThe part of central nervous system that is contained within the skull (cranium). Arising from the neural tube, the embryonic brain is comprised of three major parts including prosencephalon (the forebrain); mesencephalon (the midbrain); and rhombencephalon (the hindbrain). The developed brain consists of cerebrum; cerebellum; and other structures in the brain stem.Nervous System: Anatomy, Structure, and Classification and nervous systemNervous systemThe nervous system is a small and complex system that consists of an intricate network of neural cells (or neurons) and even more glial cells (for support and insulation). It is divided according to its anatomical components as well as its functional characteristics. The brain and spinal cord are referred to as the central nervous system, and the branches of nerves from these structures are referred to as the peripheral nervous system.Nervous System: Anatomy, Structure, and Classification.
Neuroepithelial tumors are those involving the brainBrainThe part of central nervous system that is contained within the skull (cranium). Arising from the neural tube, the embryonic brain is comprised of three major parts including prosencephalon (the forebrain); mesencephalon (the midbrain); and rhombencephalon (the hindbrain). The developed brain consists of cerebrum; cerebellum; and other structures in the brain stem.Nervous System: Anatomy, Structure, and Classification parenchyma.
Astrocytomas are graded, but not staged.
Classification of nervous systemNervous systemThe nervous system is a small and complex system that consists of an intricate network of neural cells (or neurons) and even more glial cells (for support and insulation). It is divided according to its anatomical components as well as its functional characteristics. The brain and spinal cord are referred to as the central nervous system, and the branches of nerves from these structures are referred to as the peripheral nervous system.Nervous System: Anatomy, Structure, and Classification tumors
Table: Classification of nervous systemNervous systemThe nervous system is a small and complex system that consists of an intricate network of neural cells (or neurons) and even more glial cells (for support and insulation). It is divided according to its anatomical components as well as its functional characteristics. The brain and spinal cord are referred to as the central nervous system, and the branches of nerves from these structures are referred to as the peripheral nervous system.Nervous System: Anatomy, Structure, and Classification tumors
Categories
Specific tumors
Neuroepithelial (glial & related) tumors
Pilocytic astrocytoma (grade 1)
IDH‑mutant astrocytoma (grades 2–4)
IDH‑wildtype glioblastoma (grade 4)
OligodendrogliomaOligodendrogliomaOligodendrogliomas are malignant CNS tumors arising from neural glial cell precursors. Oligodendrogliomas often arise in the frontal lobes of the brain and have a generally favorable prognosis when compared to other gliomas. Oligodendrogliomas are the 3rd most common CNS tumor. The most frequent presenting symptom is a seizure.Oligodendroglioma
EpendymomaEpendymomaEpendymomas are glial cell tumors arising from CSF-producing ependymal cells lining the ventricular system. Ependymomas most commonly occur within the posterior fossa in contact with the 4th ventricle, or within the intramedullary spinal cord. Ependymoma and choroid-plexus tumors
Embryonal tumors (e.g., medulloblastomaMedulloblastomaMedulloblastomas are malignant primitive neuroectodermal tumors that arise in the posterior fossa in children. Medulloblastomas are the most common malignant brain tumors in children. Patients with medulloblastoma present with symptoms of increased intracranial pressure and cerebellar signs, which both evolve and worsen over weeks to a few months. Medulloblastoma)
Meningeal tumors
Meningiomas
Vascular tumors
HemangioblastomasHemangioblastomasA benign tumor of the nervous system that may occur sporadically or in association with von Hippel-Lindau disease. It accounts for approximately 2% of intracranial tumors, arising most frequently in the cerebellar hemispheres and vermis. Histologically, the tumors are composed of multiple capillary and sinusoidal channels lined with endothelial cells and clusters of lipid-laden pseudoxanthoma cells. Usually solitary, these tumors can be multiple and may also occur in the brain stem, spinal cord, retina, and supratentorial compartment. Cerebellar hemangioblastomas usually present in the third decade with intracranial hypertension, and ataxia.Von Hippel-Lindau Disease
Sellar region tumors
CraniopharyngiomaCraniopharyngiomaCraniopharyngiomas are rare squamous epithelial tumors with a solid and/or cystic structure that arise from the remnants of Rathke’s pouch along the pituitary stalk, in the suprasellar region. Craniopharyngiomas are histologically benign but tend to invade surrounding structures; thus, they should be treated as low-grade malignancies.Craniopharyngioma
PituitaryPituitaryA small, unpaired gland situated in the sella turcica. It is connected to the hypothalamus by a short stalk which is called the infundibulum.Hormones: Overview and Types adenoma
Primary CNS lymphomaLymphomaA general term for various neoplastic diseases of the lymphoid tissue.Imaging of the Mediastinum (usually diffuse large B‑cell type)
Metastatic brainBrainThe part of central nervous system that is contained within the skull (cranium). Arising from the neural tube, the embryonic brain is comprised of three major parts including prosencephalon (the forebrain); mesencephalon (the midbrain); and rhombencephalon (the hindbrain). The developed brain consists of cerebrum; cerebellum; and other structures in the brain stem.Nervous System: Anatomy, Structure, and Classification tumors
Most commonly arising from:
Lung carcinomas
Breast carcinomas
Melanomas
Renal cell carcinomas
Colorectal carcinomas
Peripheral nervous systemPeripheral nervous systemThe nervous system outside of the brain and spinal cord. The peripheral nervous system has autonomic and somatic divisions. The autonomic nervous system includes the enteric, parasympathetic, and sympathetic subdivisions. The somatic nervous system includes the cranial and spinal nerves and their ganglia and the peripheral sensory receptors.Nervous System: Anatomy, Structure, and Classification tumors
Schwannomas (vestibular schwannomaVestibular schwannomaAcoustic neuroma, also referred to as vestibular schwannoma, is a benign tumor arising from Schwann cells of the vestibular component of the cranial nerve VIII. Acoustic neuroma forms within the internal auditory meatus and extends into the cerebellopontine angle. Acoustic Neuroma)
Neurofibromas
Malignant peripheral nerve sheath tumors
Classification of astrocytomas
According to WHO CNS 5, astrocytomas are classified by an integrated histologic and molecular approach, with tumorTumorInflammation grade (1–4) determined by features such as nuclear atypiaAtypiaFibrocystic Change, mitotic activity, microvascular proliferation, and necrosisNecrosisThe death of cells in an organ or tissue due to disease, injury or failure of the blood supply.Ischemic Cell Damage.
Cerebellar location → 4th‑ventricle compressionCompressionBlunt Chest Trauma/ataxiaAtaxiaImpairment of the ability to perform smoothly coordinated voluntary movements. This condition may affect the limbs, trunk, eyes, pharynx, larynx, and other structures. Ataxia may result from impaired sensory or motor function. Sensory ataxia may result from posterior column injury or peripheral nerve diseases. Motor ataxia may be associated with cerebellar diseases; cerebral cortex diseases; thalamic diseases; basal ganglia diseases; injury to the red nucleus; and other conditions.Ataxia-telangiectasia
Superficial temporal lobeTemporal lobeLower lateral part of the cerebral hemisphere responsible for auditory, olfactory, and semantic processing. It is located inferior to the lateral fissure and anterior to the occipital lobe.Cerebral Cortex: AnatomymassMassThree-dimensional lesion that occupies a space within the breastImaging of the Breast
Presents with seizuresSeizuresA seizure is abnormal electrical activity of the neurons in the cerebral cortex that can manifest in numerous ways depending on the region of the brain affected. Seizures consist of a sudden imbalance that occurs between the excitatory and inhibitory signals in cortical neurons, creating a net excitation. The 2 major classes of seizures are focal and generalized. Seizures
Anaplastic form shows ↑ mitotic activity
Subependymal giant cell astrocytoma (grade 1):
Periventricular in tuberous sclerosisSclerosisA pathological process consisting of hardening or fibrosis of an anatomical structure, often a vessel or a nerve.Wilms Tumor
Can obstruct CSF outflow → hydrocephalusHydrocephalusExcessive accumulation of cerebrospinal fluid within the cranium which may be associated with dilation of cerebral ventricles, intracranial.Subarachnoid Hemorrhage
No necrosisNecrosisThe death of cells in an organ or tissue due to disease, injury or failure of the blood supply.Ischemic Cell Damage or microvascular proliferation
Astrocytoma, IDH-mutant (grade 4):
↑ Nuclear atypiaAtypiaFibrocystic Change + mitotic activity + necrosisNecrosisThe death of cells in an organ or tissue due to disease, injury or failure of the blood supply.Ischemic Cell Damage and/or microvascular proliferation
Not called glioblastoma if IDH-mutant
Glioblastoma, IDH‑wildtype (grade 4):
NecrosisNecrosisThe death of cells in an organ or tissue due to disease, injury or failure of the blood supply.Ischemic Cell Damage and/or microvascular proliferation
“Primary” in adults (~90%) vs. “secondary” progression from lower grade (~10%)
Median survival ~12–15 months
Epidemiology
Pediatric distribution:
Gliomas account for ~50% of CNS tumors in children and adolescents; circumscribed astrocytomas are the most common.
Pilocytic astrocytoma incidenceIncidenceThe number of new cases of a given disease during a given period in a specified population. It also is used for the rate at which new events occur in a defined population. It is differentiated from prevalence, which refers to all cases in the population at a given time.Measures of Disease Frequency is ~1 per 100,000 children
Astrocytic tumors are ~31% of childhood CNS neoplasmsNeoplasmsNew abnormal growth of tissue. Malignant neoplasms show a greater degree of anaplasia and have the properties of invasion and metastasis, compared to benign neoplasms.Benign Bone Tumors.
Adult distribution:
Metastatic brainBrainThe part of central nervous system that is contained within the skull (cranium). Arising from the neural tube, the embryonic brain is comprised of three major parts including prosencephalon (the forebrain); mesencephalon (the midbrain); and rhombencephalon (the hindbrain). The developed brain consists of cerebrum; cerebellum; and other structures in the brain stem.Nervous System: Anatomy, Structure, and Classification tumors outnumber primary CNS tumors
Among primary glial tumors, diffuse astrocytomas (IDH‑mutant and IDH‑wildtype) and glioblastoma are most common.
Overall incidenceIncidenceThe number of new cases of a given disease during a given period in a specified population. It also is used for the rate at which new events occur in a defined population. It is differentiated from prevalence, which refers to all cases in the population at a given time.Measures of Disease Frequency:
All astrocytic tumors combined: ~5–6 per 100,000 person‑years
Glioblastoma, IDH‑wildtype: ~3.2 per 100,000
~13,000–15,000 new astrocytic tumors/year in the U.S.
SexSexThe totality of characteristics of reproductive structure, functions, phenotype, and genotype, differentiating the male from the female organism.Gender Dysphoria predominance:
Pilocytic astrocytoma: no clear biasBiasEpidemiological studies are designed to evaluate a hypothesized relationship between an exposure and an outcome; however, the existence and/or magnitude of these relationships may be erroneously affected by the design and execution of the study itself or by conscious or unconscious errors perpetrated by the investigators or the subjects. These systematic errors are called biases. Types of Biases
Astrocytomas occur more frequently in the following genetic disorders:
Li–Fraumeni syndrome (p53 germline mutations)
Turcot syndromeTurcot syndromeFamilial Adenomatous Polyposis (mutations in several tumorTumorInflammation suppressor genesGenesA category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms.DNA Types and Structure, including APCAPCA polyposis syndrome due to an autosomal dominant mutation of the apc genes on chromosome 5. The syndrome is characterized by the development of hundreds of adenomatous polyps in the colon and rectum of affected individuals by early adulthood.Familial Adenomatous Polyposis and MMRMMRA DNA repair pathway involved in correction of errors introduced during DNA replication when an incorrect base, which cannot form hydrogen bonds with the corresponding base in the parent strand, is incorporated into the daughter strand. Excinucleases recognize the base pair mismatch and cause a segment of polynucleotide chain to be excised from the daughter strand, thereby removing the mismatched base.Lynch syndrome)
Neurofibromatosis type 1Type 1Spinal Muscular Atrophy (NF1NF1Neurofibromatosis type 1 (NF1), also known as phakomatosis, is a neurocutaneous disorder that is most commonly of autosomal dominant inheritance due to mutations in the NF1 gene. Neurofibromatosis type 1 presents a range of clinical manifestations with the most prominent features being various pigmented skin lesions called café au lait macules (CALMs), neurofibromas, freckling of the inguinal and axillary regions, and iris hamartomas.Neurofibromatosis Type 1)
Tuberous sclerosisSclerosisA pathological process consisting of hardening or fibrosis of an anatomical structure, often a vessel or a nerve.Wilms Tumor
Ionizing radiationRadiationEmission or propagation of acoustic waves (sound), electromagnetic energy waves (such as light; radio waves; gamma rays; or x-rays), or a stream of subatomic particles (such as electrons; neutrons; protons; or alpha particles).Osteosarcoma is an established risk factor:
PituitaryPituitaryA small, unpaired gland situated in the sella turcica. It is connected to the hypothalamus by a short stalk which is called the infundibulum.Hormones: Overview and Types adenoma radiationRadiationEmission or propagation of acoustic waves (sound), electromagnetic energy waves (such as light; radio waves; gamma rays; or x-rays), or a stream of subatomic particles (such as electrons; neutrons; protons; or alpha particles).Osteosarcoma therapy: 16× increased risk of astrocytoma
Children who receive radiationRadiationEmission or propagation of acoustic waves (sound), electromagnetic energy waves (such as light; radio waves; gamma rays; or x-rays), or a stream of subatomic particles (such as electrons; neutrons; protons; or alpha particles).Osteosarcoma for acute lymphocytic leukemia (ALL): 22× increased risk of developing a CNS malignancyMalignancyHemothorax in 5–10 years (including grade 2, 3, and 4 astrocytomas)
Mutations in isocitrate dehydrogenase 1Isocitrate Dehydrogenase 1Oligodendroglioma (IDH1geneGeneA category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms.Basic Terms of Genetics):
IDH1:
Catalyzes the reversible oxidative decarboxylationDecarboxylationThe removal of a carboxyl group, usually in the form of carbon dioxide, from a chemical compound.Catabolism of Amino Acids of isocitrate → α-ketoglutarate (α-KG) in the tricarboxylic acid (TCA) cycle
Primary producer of nicotinamide adenine dinucleotideNicotinamide adenine dinucleotideA coenzyme composed of ribosylnicotinamide 5′-diphosphate coupled to adenosine 5′-phosphate by pyrophosphate linkage. It is found widely in nature and is involved in numerous enzymatic reactions in which it serves as an electron carrier by being alternately oxidized (NAD+) and reduced (NADH).Pentose Phosphate PathwayphosphatePhosphateInorganic salts of phosphoric acid.Electrolytes (NADPHNADPHNicotinamide adenine dinucleotide phosphate. A coenzyme composed of ribosylnicotinamide 5′-phosphate (nmn) coupled by pyrophosphate linkage to the 5′-phosphate adenosine 2.Pentose Phosphate Pathway) in most tissues, especially the brainBrainThe part of central nervous system that is contained within the skull (cranium). Arising from the neural tube, the embryonic brain is comprised of three major parts including prosencephalon (the forebrain); mesencephalon (the midbrain); and rhombencephalon (the hindbrain). The developed brain consists of cerebrum; cerebellum; and other structures in the brain stem.Nervous System: Anatomy, Structure, and Classification
Also involved in mitigating oxidative damage
Mutations lead to production and buildup of a 2-hydroxyglutarate (2-HG):
2-HG inhibits enzymatic function of α-ketoglutarate (KG)-dependent dioxygenases, which are involved in DNADNAA deoxyribonucleotide polymer that is the primary genetic material of all cells. Eukaryotic and prokaryotic organisms normally contain DNA in a double-stranded state, yet several important biological processes transiently involve single-stranded regions. DNA, which consists of a polysugar-phosphate backbone possessing projections of purines (adenine and guanine) and pyrimidines (thymine and cytosine), forms a double helix that is held together by hydrogen bonds between these purines and pyrimidines (adenine to thymine and guanine to cytosine).DNA Types and Structure demethylation.
↑ 2-HG causes epigenetic dysregulation → can lead to tumorTumorInflammation development
MethylationMethylationAddition of methyl groups. In histo-chemistry methylation is used to esterify carboxyl groups and remove sulfate groups by treating tissue sections with hot methanol in the presence of hydrochloric acid. .Glucocorticoids (i.e., silencing) of the methylguanine methyltransferase (MGMT) promoter:
MGMT is an enzyme involved in DNADNAA deoxyribonucleotide polymer that is the primary genetic material of all cells. Eukaryotic and prokaryotic organisms normally contain DNA in a double-stranded state, yet several important biological processes transiently involve single-stranded regions. DNA, which consists of a polysugar-phosphate backbone possessing projections of purines (adenine and guanine) and pyrimidines (thymine and cytosine), forms a double helix that is held together by hydrogen bonds between these purines and pyrimidines (adenine to thymine and guanine to cytosine).DNA Types and Structure repair (including DNADNAA deoxyribonucleotide polymer that is the primary genetic material of all cells. Eukaryotic and prokaryotic organisms normally contain DNA in a double-stranded state, yet several important biological processes transiently involve single-stranded regions. DNA, which consists of a polysugar-phosphate backbone possessing projections of purines (adenine and guanine) and pyrimidines (thymine and cytosine), forms a double helix that is held together by hydrogen bonds between these purines and pyrimidines (adenine to thymine and guanine to cytosine).DNA Types and Structure repair after chemotherapyChemotherapyOsteosarcoma with an alkylating agentAlkylating AgentAlkylating agents are cell cycle-independent antineoplastic drugs that work primarily by binding alkyl groups to various parts of DNA. The overall action produces cross-linking of DNA, leading to inhibition of DNA replication and DNA damage. Alkylating Agents and Platinum).
MGMT methylationMethylationAddition of methyl groups. In histo-chemistry methylation is used to esterify carboxyl groups and remove sulfate groups by treating tissue sections with hot methanol in the presence of hydrochloric acid. .Glucocorticoids at the promoter region:
Silences expression of the geneGeneA category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms.Basic Terms of Genetics
May occur during tumorTumorInflammation development → prevents repair of DNA damageDNA DamageInjuries to DNA that introduce deviations from its normal, intact structure and which may, if left unrepaired, result in a mutation or a block of DNA replication. These deviations may be caused by physical or chemical agents and occur by natural or unnatural, introduced circumstances. They include the introduction of illegitimate bases during replication or by deamination or other modification of bases; the loss of a base from the DNA backbone leaving an abasic site; single-strand breaks; double strand breaks; and intrastrand (pyrimidine dimers) or interstrand crosslinking. Damage can often be repaired (DNA repair). If the damage is extensive, it can induce apoptosis.DNA Repair Mechanisms
Improves chemotherapyChemotherapyOsteosarcoma responsiveness and overall survival (independent of other risk factors)
Inactivating p53 mutations (a defining co-mutation in the astrocytoma, IDH-mutant lineage)
Overexpression of platelet-derived growth factorPlatelet-derived growth factorMitogenic peptide growth hormone carried in the alpha-granules of platelets. It is released when platelets adhere to traumatized tissues. Connective tissue cells near the traumatized region respond by initiating the process of replication.Hypertrophic and Keloid Scars alpha (PDGF-A)
Certain human leukocyte antigenAntigenSubstances that are recognized by the immune system and induce an immune reaction.Vaccination (HLA) types are associated with either decreased or increased risk.
Pathophysiology
Astrocytomas usually arise in the cerebral hemispheres (i.e., parenchyma).
Regional effects on brainBrainThe part of central nervous system that is contained within the skull (cranium). Arising from the neural tube, the embryonic brain is comprised of three major parts including prosencephalon (the forebrain); mesencephalon (the midbrain); and rhombencephalon (the hindbrain). The developed brain consists of cerebrum; cerebellum; and other structures in the brain stem.Nervous System: Anatomy, Structure, and Classification parenchyma include:
↑ Intracranial pressureIntracranial PressureIdiopathic Intracranial Hypertension (ICPICPNormal intracranial pressure (ICP) is defined as < 15 mm Hg, whereas pathologically increased ICP is any pressure ≥ 20 mm Hg. Increased ICP may result from several etiologies, including trauma, intracranial hemorrhage, mass lesions, cerebral edema, increased CSF production, and decreased CSF absorption.Increased Intracranial Pressure (ICP)) may be due to:
Direct massMassThree-dimensional lesion that occupies a space within the breastImaging of the Breast effect
EdemaEdemaEdema is a condition in which excess serous fluid accumulates in the body cavity or interstitial space of connective tissues. Edema is a symptom observed in several medical conditions. It can be categorized into 2 types, namely, peripheral (in the extremities) and internal (in an organ or body cavity). Edema in surrounding brainBrainThe part of central nervous system that is contained within the skull (cranium). Arising from the neural tube, the embryonic brain is comprised of three major parts including prosencephalon (the forebrain); mesencephalon (the midbrain); and rhombencephalon (the hindbrain). The developed brain consists of cerebrum; cerebellum; and other structures in the brain stem.Nervous System: Anatomy, Structure, and Classification tissue
↑ Blood volume
↑ CSF volume/hydrocephalusHydrocephalusExcessive accumulation of cerebrospinal fluid within the cranium which may be associated with dilation of cerebral ventricles, intracranial.Subarachnoid Hemorrhage
Disruption of normal parenchymal functions are due to:
Free radicalsFree radicalsHighly reactive molecules with an unsatisfied electron valence pair. Free radicals are produced in both normal and pathological processes. They are proven or suspected agents of tissue damage in a wide variety of circumstances including radiation, damage from environment chemicals, and aging. Natural and pharmacological prevention of free radical damage is being actively investigated.Ischemic Cell Damage
Altered electrolytesElectrolytesElectrolytes are mineral salts that dissolve in water and dissociate into charged particles called ions, which can be either be positively (cations) or negatively (anions) charged. Electrolytes are distributed in the extracellular and intracellular compartments in different concentrations. Electrolytes are essential for various basic life-sustaining functions.Electrolytes
Neurotransmitters
Release and recruitmentRecruitmentSkeletal Muscle Contraction of cellular mediators (e.g., cytokinesCytokinesNon-antibody proteins secreted by inflammatory leukocytes and some non-leukocytic cells, that act as intercellular mediators. They differ from classical hormones in that they are produced by a number of tissue or cell types rather than by specialized glands. They generally act locally in a paracrine or autocrine rather than endocrine manner.Adaptive Immune Response)
Clinical Presentation
Neurologic signs and symptoms related to astrocytomas result from perturbations in CNS function.
Onset of symptoms
Grades 1 and 2: Onset is usually subtle and tumors develop slowly because of the brainBrainThe part of central nervous system that is contained within the skull (cranium). Arising from the neural tube, the embryonic brain is comprised of three major parts including prosencephalon (the forebrain); mesencephalon (the midbrain); and rhombencephalon (the hindbrain). The developed brain consists of cerebrum; cerebellum; and other structures in the brain stem.Nervous System: Anatomy, Structure, and Classification’s ability to temporarily adapt to the presence of a slow-growing tumorTumorInflammation.
Grades 3 and 4:Onset is more often sudden and/or debilitating.
Symptoms
Symptoms are primarily dependent on the location of the tumorTumorInflammation in the brainBrainThe part of central nervous system that is contained within the skull (cranium). Arising from the neural tube, the embryonic brain is comprised of three major parts including prosencephalon (the forebrain); mesencephalon (the midbrain); and rhombencephalon (the hindbrain). The developed brain consists of cerebrum; cerebellum; and other structures in the brain stem.Nervous System: Anatomy, Structure, and Classification The symptoms are divided into 2 categories:
General symptoms: symptoms that can occur with tumors in any location
HeadacheHeadacheThe symptom of pain in the cranial region. It may be an isolated benign occurrence or manifestation of a wide variety of headache disorders.Brain Abscess (usually in the morning)
NauseaNauseaAn unpleasant sensation in the stomach usually accompanied by the urge to vomit. Common causes are early pregnancy, sea and motion sickness, emotional stress, intense pain, food poisoning, and various enteroviruses.Antiemetics and/or vomitingVomitingThe forcible expulsion of the contents of the stomach through the mouth.Hypokalemia
Cognitive difficulties:
MemoryMemoryComplex mental function having four distinct phases: (1) memorizing or learning, (2) retention, (3) recall, and (4) recognition. Clinically, it is usually subdivided into immediate, recent, and remote memory.Psychiatric Assessment problems
AtaxiaAtaxiaImpairment of the ability to perform smoothly coordinated voluntary movements. This condition may affect the limbs, trunk, eyes, pharynx, larynx, and other structures. Ataxia may result from impaired sensory or motor function. Sensory ataxia may result from posterior column injury or peripheral nerve diseases. Motor ataxia may be associated with cerebellar diseases; cerebral cortex diseases; thalamic diseases; basal ganglia diseases; injury to the red nucleus; and other conditions.Ataxia-telangiectasia
PapilledemaPapilledemaSwelling of the optic disk, usually in association with increased intracranial pressure, characterized by hyperemia, blurring of the disk margins, microhemorrhages, blind spot enlargement, and engorgement of retinal veins. Chronic papilledema may cause optic atrophy and visual loss.Idiopathic Intracranial Hypertension
Focal symptoms: symptoms that occur because of tumors in specific locations
SeizuresSeizuresA seizure is abnormal electrical activity of the neurons in the cerebral cortex that can manifest in numerous ways depending on the region of the brain affected. Seizures consist of a sudden imbalance that occurs between the excitatory and inhibitory signals in cortical neurons, creating a net excitation. The 2 major classes of seizures are focal and generalized. Seizures:
Most common symptom of astrocytomas
Low-grade tumors are more likely to cause seizuresSeizuresA seizure is abnormal electrical activity of the neurons in the cerebral cortex that can manifest in numerous ways depending on the region of the brain affected. Seizures consist of a sudden imbalance that occurs between the excitatory and inhibitory signals in cortical neurons, creating a net excitation. The 2 major classes of seizures are focal and generalized. Seizures than high-grade tumors.
AphasiaAphasiaA cognitive disorder marked by an impaired ability to comprehend or express language in its written or spoken form. This condition is caused by diseases which affect the language areas of the dominant hemisphere. Clinical features are used to classify the various subtypes of this condition. General categories include receptive, expressive, and mixed forms of aphasia.Ischemic Stroke
NeuroimagingNeuroimagingNon-invasive methods of visualizing the central nervous system, especially the brain, by various imaging modalities.Febrile Infant is essential for detection and initial characterization, but definitive diagnosis and gradingGradingMethods which attempt to express in replicable terms the level of cell differentiation in neoplasms as increasing anaplasia correlates with the aggressiveness of the neoplasm.Grading, Staging, and Metastasis require histopathology and molecular testing.
MRI
Gold standard imaging study
Preferably ordered with and without gadoliniumGadoliniumAn element of the rare earth family of metals. It has the atomic symbol gd, atomic number 64, and atomic weight 157. 25. Its oxide is used in the control rods of some nuclear reactors.Magnetic Resonance Imaging (MRI) enhancement
Low-grade astrocytomas typically do not enhance with contrast.
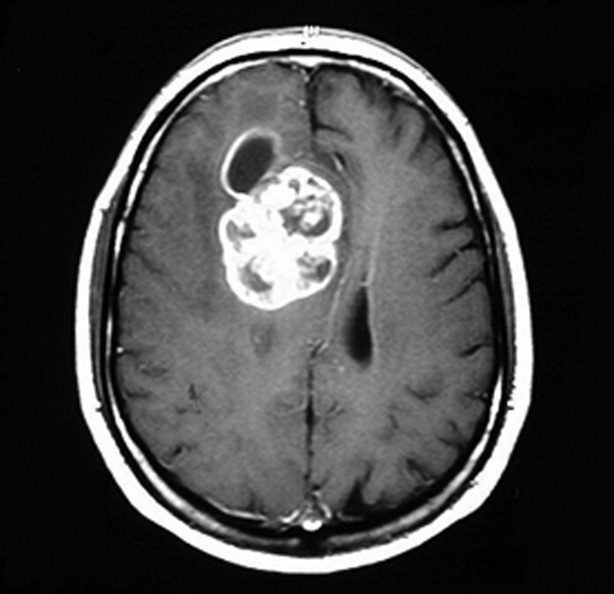
Contrast MRI showing glioblastoma multiforme
Image: “Glioblastoma multiforme” by Duncan JS, de Tisi J. License: CC BY 3.0
CT scan
Indications:
Contraindication to MRI
Acute settings (e.g., where hemorrhage and/or stroke needs to be ruled out)
CT of the chest/abdomen/pelvisPelvisThe pelvis consists of the bony pelvic girdle, the muscular and ligamentous pelvic floor, and the pelvic cavity, which contains viscera, vessels, and multiple nerves and muscles. The pelvic girdle, composed of 2 “hip” bones and the sacrum, is a ring-like bony structure of the axial skeleton that links the vertebral column with the lower extremities.Pelvis: Anatomy may be warranted to look for alternative primary lesions if metastasisMetastasisThe transfer of a neoplasm from one organ or part of the body to another remote from the primary site.Grading, Staging, and Metastasis is suspected.
Findings show a poorly defined parenchymal massMassThree-dimensional lesion that occupies a space within the breastImaging of the Breast.
Low-grade lesions typically do not enhance with contrast.
Other tests
EEGEEGSeizures: may be used to evaluate and monitor seizure activity
CSF studies: may assist in ruling out other diagnoses (e.g., CNS lymphomaLymphomaA general term for various neoplastic diseases of the lymphoid tissue.Imaging of the Mediastinum, metastasisMetastasisThe transfer of a neoplasm from one organ or part of the body to another remote from the primary site.Grading, Staging, and Metastasis)
BiopsyBiopsyRemoval and pathologic examination of specimens from the living body.Ewing Sarcoma
Tissue (via resection or stereotactic biopsyBiopsyRemoval and pathologic examination of specimens from the living body.Ewing Sarcoma) is required for histopathologic and molecular classification. A biopsyBiopsyRemoval and pathologic examination of specimens from the living body.Ewing Sarcoma is indicated for gradingGradingMethods which attempt to express in replicable terms the level of cell differentiation in neoplasms as increasing anaplasia correlates with the aggressiveness of the neoplasm.Grading, Staging, and Metastasis and/or confirming the diagnosis after the tumorTumorInflammation is identified on neuroimagingNeuroimagingNon-invasive methods of visualizing the central nervous system, especially the brain, by various imaging modalities.Febrile Infant.
Mild to moderate increase in number of glial cell nuclei
Nuclear pleomorphism
NucleusNucleusWithin a eukaryotic cell, a membrane-limited body which contains chromosomes and one or more nucleoli (cell nucleolus). The nuclear membrane consists of a double unit-type membrane which is perforated by a number of pores; the outermost membrane is continuous with the endoplasmic reticulum. A cell may contain more than one nucleus.The Cell: Organelles:cytoplasm ratio quite high
Intervening feltwork of fine, glial fibrillary acidic protein (GFAP) astrocytic cell processes
Grossly: poorly defined tumors that are gray in color
Increased mitotic figures, cellularity, and nuclear pleomorphism
Vascular and/or endothelial cell proliferation
Areas of necrosisNecrosisThe death of cells in an organ or tissue due to disease, injury or failure of the blood supply.Ischemic Cell Damage (grossly appear firm and white or soft and yellow)
Pilocytic astrocytoma with prominent endothelial cells as well as neoplastic cells harboring mitotic figures and bland, oval nuclei
Image: “Hemorrhagic Pilocytic Astrocytomas in Adults: A Case Report and Literature Review” by Galgano MA, Padalino DJ, Fullmer J, Krishnamurthy S. License: CC BY 3.0
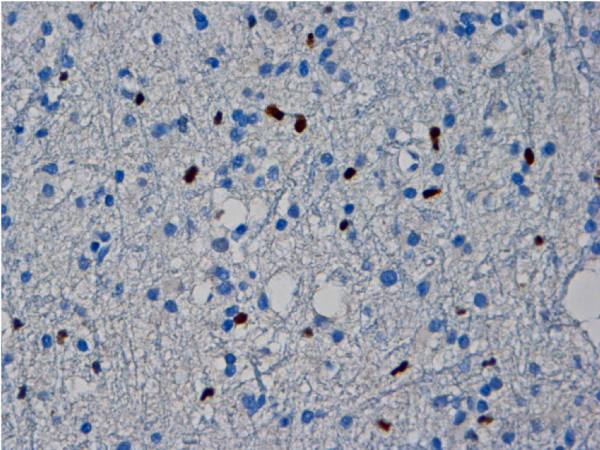
Image of a minichromosome maintenance protein 2 (MCM2) immunostaining of a grade II astrocytomas with positive neoplastic astrocytic cell nuclei (dark) (40× objective)
Image: “Expression and clinical significance of the proliferation marker minichromosome maintenance protein 2 (Mcm2) in diffuse astrocytomas WHO grade II” by Lind-Landström T, Varughese RK, Sundstrøm S, Torp SH. License: CC BY 2.0
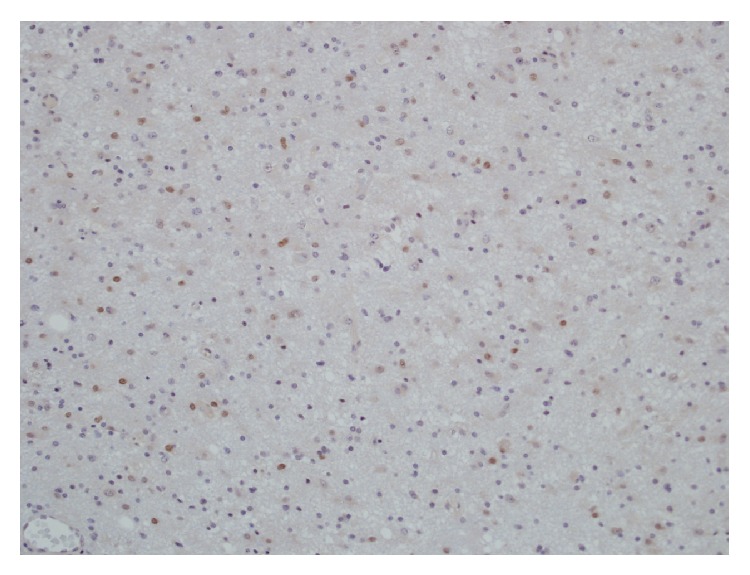
Microphotograph demonstrating significant p53 immunopositivity of anaplastic astrocytoma
Image: “Malignant trigeminal nerve sheath tumor and anaplastic astrocytoma collision tumor with high proliferative activity and tumor suppressor p53 expression” by Kurdi M, Al-Ardati H, Baeesa SS. License: Public Domain
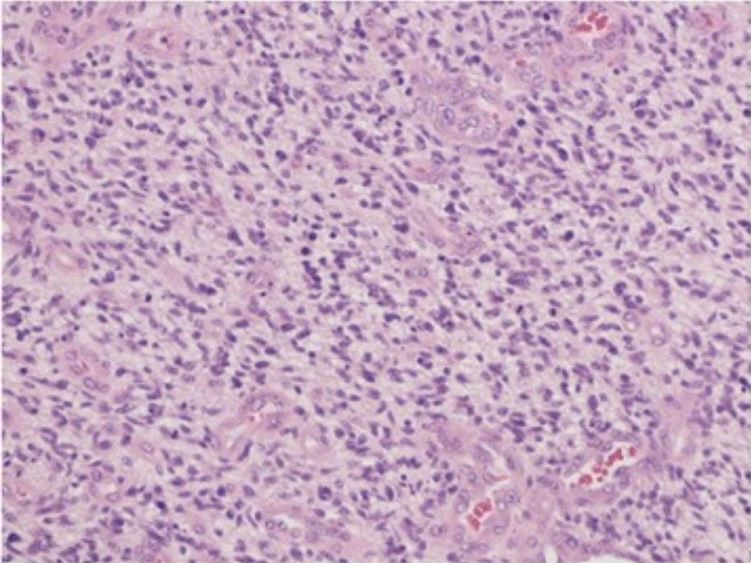
Microphotography revealing highly cellular malignant glial neoplasm, embedded in glial fibrillary background, with mitosis, endothelial proliferation, and necrosis Findings are consistent with glioblastoma (hematoxylin–eosin stain, ×40)
Image: “De novo glioblastoma in the territory of a recent middle cerebral artery infarction and a residual meningioma: pathogenesis revisited” by Yaghmour W, Kurdi ME, Baeesa SS. License: CC BY 4.0
Management and Prognosis
Management by grade
Management depends on the grade, location of the tumorTumorInflammation (i.e., how much can be safely resected), and symptoms. Management typically involves a combination of surgery, radiationRadiationEmission or propagation of acoustic waves (sound), electromagnetic energy waves (such as light; radio waves; gamma rays; or x-rays), or a stream of subatomic particles (such as electrons; neutrons; protons; or alpha particles).Osteosarcoma, and chemotherapyChemotherapyOsteosarcoma.
MorbidityMorbidityThe proportion of patients with a particular disease during a given year per given unit of population.Measures of Health Status associated with the treatments
Relatively young age of the typical patient
Consider maximal safe surgical resection if tumorTumorInflammation is accessible.
Partial surgical removal when the tumorTumorInflammation involves crucial parts of the brainBrainThe part of central nervous system that is contained within the skull (cranium). Arising from the neural tube, the embryonic brain is comprised of three major parts including prosencephalon (the forebrain); mesencephalon (the midbrain); and rhombencephalon (the hindbrain). The developed brain consists of cerebrum; cerebellum; and other structures in the brain stem.Nervous System: Anatomy, Structure, and Classification
Consider radiationRadiationEmission or propagation of acoustic waves (sound), electromagnetic energy waves (such as light; radio waves; gamma rays; or x-rays), or a stream of subatomic particles (such as electrons; neutrons; protons; or alpha particles).Osteosarcoma therapy when the tumorTumorInflammation is not fully resectable.
Astrocytoma, IDH‑mutant (grade 2):
Surgical resection is recommended for accessible tumors:
Not curative
Additional therapies (i.e., radiationRadiationEmission or propagation of acoustic waves (sound), electromagnetic energy waves (such as light; radio waves; gamma rays; or x-rays), or a stream of subatomic particles (such as electrons; neutrons; protons; or alpha particles).Osteosarcoma therapy and chemotherapyChemotherapyOsteosarcoma) are ultimately required in all patientsPatientsIndividuals participating in the health care system for the purpose of receiving therapeutic, diagnostic, or preventive procedures.Clinician–Patient Relationship.
RadiationRadiationEmission or propagation of acoustic waves (sound), electromagnetic energy waves (such as light; radio waves; gamma rays; or x-rays), or a stream of subatomic particles (such as electrons; neutrons; protons; or alpha particles).Osteosarcoma plus adjuvantAdjuvantSubstances that augment, stimulate, activate, potentiate, or modulate the immune response at either the cellular or humoral level. The classical agents (freund’s adjuvant, bcg, corynebacterium parvum, et al.) contain bacterial antigens. Some are endogenous (e.g., histamine, interferon, transfer factor, tuftsin, interleukin-1). Their mode of action is either non-specific, resulting in increased immune responsiveness to a wide variety of antigens, or antigen-specific, i.e., affecting a restricted type of immune response to a narrow group of antigens. The therapeutic efficacy of many biological response modifiers is related to their antigen-specific immunoadjuvanticity.VaccinationchemotherapyChemotherapyOsteosarcoma (typically temozolomideTemozolomideA dacarbazine derivative that is used as an alkylating antineoplastic agent for the treatment of malignant glioma and malignant melanoma.Alkylating Agents and Platinum)
Follow-up to check for recurrence/progression
Grades 3 and 4:
Standard therapy is surgery, radiationRadiationEmission or propagation of acoustic waves (sound), electromagnetic energy waves (such as light; radio waves; gamma rays; or x-rays), or a stream of subatomic particles (such as electrons; neutrons; protons; or alpha particles).Osteosarcoma, and chemotherapyChemotherapyOsteosarcoma (typically with temozolomideTemozolomideA dacarbazine derivative that is used as an alkylating antineoplastic agent for the treatment of malignant glioma and malignant melanoma.Alkylating Agents and Platinum).
Surgery may include:
Resection
Symptom-relieving procedures (e.g., shunt placement to relieve hydrocephalusHydrocephalusExcessive accumulation of cerebrospinal fluid within the cranium which may be associated with dilation of cerebral ventricles, intracranial.Subarachnoid Hemorrhage)
If surgery is not possible, then radiationRadiationEmission or propagation of acoustic waves (sound), electromagnetic energy waves (such as light; radio waves; gamma rays; or x-rays), or a stream of subatomic particles (such as electrons; neutrons; protons; or alpha particles).Osteosarcoma and chemotherapyChemotherapyOsteosarcoma are used.
Other therapy to consider:
DVTDVTDeep vein thrombosis (DVT) usually occurs in the deep veins of the lower extremities. The affected veins include the femoral, popliteal, iliofemoral, and pelvic veins. Proximal DVT is more likely to cause a pulmonary embolism (PE) and is generally considered more serious. Deep Vein ThrombosisprophylaxisProphylaxisCephalosporins in nonambulatory and hospitalized patientsPatientsIndividuals participating in the health care system for the purpose of receiving therapeutic, diagnostic, or preventive procedures.Clinician–Patient Relationship
AnticonvulsantAnticonvulsantAnticonvulsant drugs are pharmacological agents used to achieve seizure control and/or prevent seizure episodes. Anticonvulsants encompass various drugs with different mechanisms of action including ion-channel (Na+ and Ca+2) blocking and GABA reuptake inhibition. First-Generation Anticonvulsant Drugs therapy in patientsPatientsIndividuals participating in the health care system for the purpose of receiving therapeutic, diagnostic, or preventive procedures.Clinician–Patient Relationship witha history of seizuresSeizuresA seizure is abnormal electrical activity of the neurons in the cerebral cortex that can manifest in numerous ways depending on the region of the brain affected. Seizures consist of a sudden imbalance that occurs between the excitatory and inhibitory signals in cortical neurons, creating a net excitation. The 2 major classes of seizures are focal and generalized. Seizures (prophylactic anticonvulsantAnticonvulsantAnticonvulsant drugs are pharmacological agents used to achieve seizure control and/or prevent seizure episodes. Anticonvulsants encompass various drugs with different mechanisms of action including ion-channel (Na+ and Ca+2) blocking and GABA reuptake inhibition. First-Generation Anticonvulsant Drugs therapy remains controversial)
PrognosisPrognosisA prediction of the probable outcome of a disease based on a individual’s condition and the usual course of the disease as seen in similar situations.Non-Hodgkin Lymphomas
Factors that affect prognosisPrognosisA prediction of the probable outcome of a disease based on a individual’s condition and the usual course of the disease as seen in similar situations.Non-Hodgkin Lymphomas include:
Use of adjuvantAdjuvantSubstances that augment, stimulate, activate, potentiate, or modulate the immune response at either the cellular or humoral level. The classical agents (freund’s adjuvant, bcg, corynebacterium parvum, et al.) contain bacterial antigens. Some are endogenous (e.g., histamine, interferon, transfer factor, tuftsin, interleukin-1). Their mode of action is either non-specific, resulting in increased immune responsiveness to a wide variety of antigens, or antigen-specific, i.e., affecting a restricted type of immune response to a narrow group of antigens. The therapeutic efficacy of many biological response modifiers is related to their antigen-specific immunoadjuvanticity.VaccinationradiationRadiationEmission or propagation of acoustic waves (sound), electromagnetic energy waves (such as light; radio waves; gamma rays; or x-rays), or a stream of subatomic particles (such as electrons; neutrons; protons; or alpha particles).Osteosarcoma therapy and/or chemotherapyChemotherapyOsteosarcoma
Age: younger age at diagnosis associated with longer survival.
Functional status (e.g., minimal symptoms and/or normal neurologic function) associated with longer survival
When a patient presents with neurologic symptoms on exam, the differential diagnosis may include vascular processes (e.g., hemorrhage, infarctInfarctArea of necrotic cells in an organ, arising mainly from hypoxia and ischemiaIschemic Cell Damage), infectionsInfectionsInvasion of the host organism by microorganisms or their toxins or by parasites that can cause pathological conditions or diseases.Chronic Granulomatous Disease (e.g., abscessAbscessAccumulation of purulent material in tissues, organs, or circumscribed spaces, usually associated with signs of infection.Chronic Granulomatous Disease, viral encephalitisViral encephalitisInflammation of brain parenchymal tissue as a result of viral infection. Encephalitis may occur as primary or secondary manifestation of togaviridae infections; herpesviridae infections; adenoviridae infections; flaviviridae infections; bunyaviridae infections; picornaviridae infections; paramyxoviridae infections; orthomyxoviridae infections; retroviridae infections; and arenaviridae infections.Encephalitis), and inflammatory processes (e.g., multiple sclerosisSclerosisA pathological process consisting of hardening or fibrosis of an anatomical structure, often a vessel or a nerve.Wilms Tumor) in addition to primary brainBrainThe part of central nervous system that is contained within the skull (cranium). Arising from the neural tube, the embryonic brain is comprised of three major parts including prosencephalon (the forebrain); mesencephalon (the midbrain); and rhombencephalon (the hindbrain). The developed brain consists of cerebrum; cerebellum; and other structures in the brain stem.Nervous System: Anatomy, Structure, and Classification tumors. Imaging is typically able to narrow the differential to a brainBrainThe part of central nervous system that is contained within the skull (cranium). Arising from the neural tube, the embryonic brain is comprised of three major parts including prosencephalon (the forebrain); mesencephalon (the midbrain); and rhombencephalon (the hindbrain). The developed brain consists of cerebrum; cerebellum; and other structures in the brain stem.Nervous System: Anatomy, Structure, and ClassificationtumorTumorInflammation.
OligodendrogliomaOligodendrogliomaOligodendrogliomas are malignant CNS tumors arising from neural glial cell precursors. Oligodendrogliomas often arise in the frontal lobes of the brain and have a generally favorable prognosis when compared to other gliomas. Oligodendrogliomas are the 3rd most common CNS tumor. The most frequent presenting symptom is a seizure.Oligodendroglioma: CNS tumorTumorInflammation arising from oligodendrocytes. OligodendrogliomaOligodendrogliomaOligodendrogliomas are malignant CNS tumors arising from neural glial cell precursors. Oligodendrogliomas often arise in the frontal lobes of the brain and have a generally favorable prognosis when compared to other gliomas. Oligodendrogliomas are the 3rd most common CNS tumor. The most frequent presenting symptom is a seizure.Oligodendroglioma most commonly develops in the cerebral hemisphere, usually in the frontal lobeFrontal lobeThe part of the cerebral hemisphere anterior to the central sulcus, and anterior and superior to the lateral sulcus.Cerebral Cortex: Anatomy. Oligodendrogliomas can present with focal neurologic deficitsNeurologic DeficitsHigh-Risk Headaches, seizuresSeizuresA seizure is abnormal electrical activity of the neurons in the cerebral cortex that can manifest in numerous ways depending on the region of the brain affected. Seizures consist of a sudden imbalance that occurs between the excitatory and inhibitory signals in cortical neurons, creating a net excitation. The 2 major classes of seizures are focal and generalized. Seizures, and personality changes depending on its exact location. Diagnosis is made by MRI and biopsyBiopsyRemoval and pathologic examination of specimens from the living body.Ewing Sarcoma. Management involves surgical resection, possibly accompanied by radiationRadiationEmission or propagation of acoustic waves (sound), electromagnetic energy waves (such as light; radio waves; gamma rays; or x-rays), or a stream of subatomic particles (such as electrons; neutrons; protons; or alpha particles).Osteosarcoma and/or chemotherapyChemotherapyOsteosarcoma.
MedulloblastomaMedulloblastomaMedulloblastomas are malignant primitive neuroectodermal tumors that arise in the posterior fossa in children. Medulloblastomas are the most common malignant brain tumors in children. Patients with medulloblastoma present with symptoms of increased intracranial pressure and cerebellar signs, which both evolve and worsen over weeks to a few months. Medulloblastoma:tumorTumorInflammation that arises in the posterior fossa. MedulloblastomaMedulloblastomaMedulloblastomas are malignant primitive neuroectodermal tumors that arise in the posterior fossa in children. Medulloblastomas are the most common malignant brain tumors in children. Patients with medulloblastoma present with symptoms of increased intracranial pressure and cerebellar signs, which both evolve and worsen over weeks to a few months. Medulloblastoma is the most common malignant brainBrainThe part of central nervous system that is contained within the skull (cranium). Arising from the neural tube, the embryonic brain is comprised of three major parts including prosencephalon (the forebrain); mesencephalon (the midbrain); and rhombencephalon (the hindbrain). The developed brain consists of cerebrum; cerebellum; and other structures in the brain stem.Nervous System: Anatomy, Structure, and ClassificationtumorTumorInflammation in children. PatientsPatientsIndividuals participating in the health care system for the purpose of receiving therapeutic, diagnostic, or preventive procedures.Clinician–Patient Relationship present with symptoms of increased ICPIncreased ICPExcessive accumulation of cerebrospinal fluid within the cranium which may be associated with dilation of cerebral ventricles, intracranial.Subarachnoid Hemorrhage as well as cerebellar signs that generally worsen over time. Diagnosis is suspected based on MRI findings, but histopathologic analysis at the time of surgical resection is required for diagnosis. Treatment is with a combination of surgery, radiationRadiationEmission or propagation of acoustic waves (sound), electromagnetic energy waves (such as light; radio waves; gamma rays; or x-rays), or a stream of subatomic particles (such as electrons; neutrons; protons; or alpha particles).Osteosarcoma therapy, and chemotherapyChemotherapyOsteosarcoma.
EpendymomaEpendymomaEpendymomas are glial cell tumors arising from CSF-producing ependymal cells lining the ventricular system. Ependymomas most commonly occur within the posterior fossa in contact with the 4th ventricle, or within the intramedullary spinal cord. Ependymoma: subset of glial tumors that often arise in or adjacent to the ependymal lining of the ventricular systemVentricular SystemThe ventricular system is an extension of the subarachnoid space into the brain consisting of a series of interconnecting spaces and channels. Four chambers are filled with cerebrospinal fluid (CSF): the paired lateral ventricles, the unpaired 3rd ventricle, and the unpaired 4th ventricle. Ventricular System: Anatomy, most commonly within the posterior fossa, in contact with the 4th ventricle, or within the intramedullary spinal cordSpinal cordThe spinal cord is the major conduction pathway connecting the brain to the body; it is part of the CNS. In cross section, the spinal cord is divided into an H-shaped area of gray matter (consisting of synapsing neuronal cell bodies) and a surrounding area of white matter (consisting of ascending and descending tracts of myelinated axons). Spinal Cord: Anatomy. The clinical presentation varies depending on the location of the tumorTumorInflammation. MRI is the standard imaging technique, but histologic confirmation is required for diagnosis. Treatment involves surgical resection and adjuvantAdjuvantSubstances that augment, stimulate, activate, potentiate, or modulate the immune response at either the cellular or humoral level. The classical agents (freund’s adjuvant, bcg, corynebacterium parvum, et al.) contain bacterial antigens. Some are endogenous (e.g., histamine, interferon, transfer factor, tuftsin, interleukin-1). Their mode of action is either non-specific, resulting in increased immune responsiveness to a wide variety of antigens, or antigen-specific, i.e., affecting a restricted type of immune response to a narrow group of antigens. The therapeutic efficacy of many biological response modifiers is related to their antigen-specific immunoadjuvanticity.VaccinationradiationRadiationEmission or propagation of acoustic waves (sound), electromagnetic energy waves (such as light; radio waves; gamma rays; or x-rays), or a stream of subatomic particles (such as electrons; neutrons; protons; or alpha particles).Osteosarcoma or chemotherapyChemotherapyOsteosarcoma (based on age).
Metastatic tumorTumorInflammation: neoplastic cells that have spread to the brainBrainThe part of central nervous system that is contained within the skull (cranium). Arising from the neural tube, the embryonic brain is comprised of three major parts including prosencephalon (the forebrain); mesencephalon (the midbrain); and rhombencephalon (the hindbrain). The developed brain consists of cerebrum; cerebellum; and other structures in the brain stem.Nervous System: Anatomy, Structure, and Classification from primary tumors elsewhere in the body. Metastatic neoplasmsNeoplasmsNew abnormal growth of tissue. Malignant neoplasms show a greater degree of anaplasia and have the properties of invasion and metastasis, compared to benign neoplasms.Benign Bone Tumors are the most common neoplasmsNeoplasmsNew abnormal growth of tissue. Malignant neoplasms show a greater degree of anaplasia and have the properties of invasion and metastasis, compared to benign neoplasms.Benign Bone Tumors in the brainBrainThe part of central nervous system that is contained within the skull (cranium). Arising from the neural tube, the embryonic brain is comprised of three major parts including prosencephalon (the forebrain); mesencephalon (the midbrain); and rhombencephalon (the hindbrain). The developed brain consists of cerebrum; cerebellum; and other structures in the brain stem.Nervous System: Anatomy, Structure, and Classification. NeuroimagingNeuroimagingNon-invasive methods of visualizing the central nervous system, especially the brain, by various imaging modalities.Febrile Infant often shows multiple foci of the carcinoma, suggesting nonbrain origin. Clinical presentation depends on the primary tumorTumorInflammation and location and extent of brainBrainThe part of central nervous system that is contained within the skull (cranium). Arising from the neural tube, the embryonic brain is comprised of three major parts including prosencephalon (the forebrain); mesencephalon (the midbrain); and rhombencephalon (the hindbrain). The developed brain consists of cerebrum; cerebellum; and other structures in the brain stem.Nervous System: Anatomy, Structure, and ClassificationmetastasisMetastasisThe transfer of a neoplasm from one organ or part of the body to another remote from the primary site.Grading, Staging, and Metastasis. Treatment is directed at the underlying neoplasm and may involve surgical resection, radiationRadiationEmission or propagation of acoustic waves (sound), electromagnetic energy waves (such as light; radio waves; gamma rays; or x-rays), or a stream of subatomic particles (such as electrons; neutrons; protons; or alpha particles).Osteosarcoma therapy, and chemotherapyChemotherapyOsteosarcoma.
Sejda, A., Grajkowska, W., Trubicka, J., Szutowicz, E., Wojdacz, T. K., Kloc, W., & Łyżka-Świeszewska, E. (2022). WHO CNS5 2021 classification of gliomas: A practical review and road signs for diagnosing pathologists and proper patho-clinical and neuro-oncological cooperation. Folia Neuropathologica, 60(2), 137–152. https://doi.org/10.5114/fn.2022.118183
Create your free account or log in to continue reading!