Amino acidsAmino acidsOrganic compounds that generally contain an amino (-NH2) and a carboxyl (-COOH) group. Twenty alpha-amino acids are the subunits which are polymerized to form proteins.Basics of Amino Acids (AAs) can be acquired through the breakdown of intracellular or ingested dietary proteinsProteinsLinear polypeptides that are synthesized on ribosomes and may be further modified, crosslinked, cleaved, or assembled into complex proteins with several subunits. The specific sequence of amino acids determines the shape the polypeptide will take, during protein folding, and the function of the protein.Energy Homeostasis. Amino acidsAmino acidsOrganic compounds that generally contain an amino (-NH2) and a carboxyl (-COOH) group. Twenty alpha-amino acids are the subunits which are polymerized to form proteins.Basics of Amino Acids can enter 3 metabolic routes within the body. They can 1) be recycled to synthesize new proteinsProteinsLinear polypeptides that are synthesized on ribosomes and may be further modified, crosslinked, cleaved, or assembled into complex proteins with several subunits. The specific sequence of amino acids determines the shape the polypeptide will take, during protein folding, and the function of the protein.Energy Homeostasis; 2) combine with cofactors and substances to create amino acidAmino acidAmino acids (AAs) are composed of a central carbon atom attached to a carboxyl group, an amino group, a hydrogen atom, and a side chain (R group). Basics of Amino Acids derivatives; or 3) be catabolized into their functional groups and carbon skeletons. This process releases ammonium, which moves into the ureaUreaA compound formed in the liver from ammonia produced by the deamination of amino acids. It is the principal end product of protein catabolism and constitutes about one half of the total urinary solids.Urea Cycle cycle and produces intermediates for energetic metabolic pathways.
Amino acidsAmino acidsOrganic compounds that generally contain an amino (-NH2) and a carboxyl (-COOH) group. Twenty alpha-amino acids are the subunits which are polymerized to form proteins.Basics of Amino Acids (AAs) follow 3 main metabolic pathways for their metabolism:
SynthesisSynthesisPolymerase Chain Reaction (PCR) of new proteinsProteinsLinear polypeptides that are synthesized on ribosomes and may be further modified, crosslinked, cleaved, or assembled into complex proteins with several subunits. The specific sequence of amino acids determines the shape the polypeptide will take, during protein folding, and the function of the protein.Energy Homeostasis
Formation of amino acidAmino acidAmino acids (AAs) are composed of a central carbon atom attached to a carboxyl group, an amino group, a hydrogen atom, and a side chain (R group). Basics of Amino Acids derivatives
Catabolism of AAs:
Catabolism consists of the breakdown of complex molecules into smaller units to produce energy or to be used in anabolic reactions.
Removal or exchange of functional groups:
Involves transamination, deamination, and decarboxylation
Releases excess nitrogenNitrogenAn element with the atomic symbol n, atomic number 7, and atomic weight [14. 00643; 14. 00728]. Nitrogen exists as a diatomic gas and makes up about 78% of the earth’s atmosphere by volume. It is a constituent of proteins and nucleic acids and found in all living cells.Urea Cycle in the form of ammonium (NH4+), which then enters the ureaUreaA compound formed in the liver from ammonia produced by the deamination of amino acids. It is the principal end product of protein catabolism and constitutes about one half of the total urinary solids.Urea Cycle cycle, is converted into ureaUreaA compound formed in the liver from ammonia produced by the deamination of amino acids. It is the principal end product of protein catabolism and constitutes about one half of the total urinary solids.Urea Cycle, and excreted through the urine
Catabolism of the remaining carbon skeleton:
In general, all 20 AAs can be broken down into 1 of 6 intermediates: pyruvatePyruvateDerivatives of pyruvic acid, including its salts and esters.Glycolysis, acetyl-CoAAcetyl-CoAAcetyl CoA participates in the biosynthesis of fatty acids and sterols, in the oxidation of fatty acids and in the metabolism of many amino acids. It also acts as a biological acetylating agent.Citric Acid Cycle, oxaloacetateOxaloacetateDerivatives of oxaloacetic acid. Included under this heading are a broad variety of acid forms, salts, esters, and amides that include a 2-keto-1, 4-carboxy aliphatic structure.Citric Acid Cycle, alpha-ketoglutarate, succinyl-CoASuccinyl-CoACitric Acid Cycle, or fumarateFumarateCitric Acid Cycle.
Ketogenic AAs metabolize to acetyl-CoAAcetyl-CoAAcetyl CoA participates in the biosynthesis of fatty acids and sterols, in the oxidation of fatty acids and in the metabolism of many amino acids. It also acts as a biological acetylating agent.Citric Acid Cycle, later used in the citric acid cycleCitric acid cycleThe citric acid cycle, also known as the tricarboxylic acid (TCA) cycle or the Krebs cycle, is a cyclic set of reactions that occurs in the mitochondrial matrix. The TCA cycle is the continuation of any metabolic pathway that produces pyruvate, which is converted into its main substrate, acetyl-CoA. Citric Acid Cycle, ketogenesisKetogenesisKetone Body Metabolism, or fatty acid synthesisSynthesisPolymerase Chain Reaction (PCR).
Glucogenic AAs are converted into glucoseGlucoseA primary source of energy for living organisms. It is naturally occurring and is found in fruits and other parts of plants in its free state. It is used therapeutically in fluid and nutrient replacement.Lactose Intolerance through gluconeogenesisGluconeogenesisGluconeogenesis is the process of making glucose from noncarbohydrate precursors. This metabolic pathway is more than just a reversal of glycolysis. Gluconeogenesis provides the body with glucose not obtained from food, such as during a fasting period. The production of glucose is critical for organs and cells that cannot use fat for fuel. Gluconeogenesis.
Some AAs are both glucogenic and ketogenic.
Schematic diagram of the metabolism of amino acids, including the 3 major pathways: reutilization in the synthesis of new proteins, union with cofactors to produce amino acid derivatives, and catabolism. Catabolism of amino acids includes the removal of functional groups and the breakdown of the carbon skeletons.
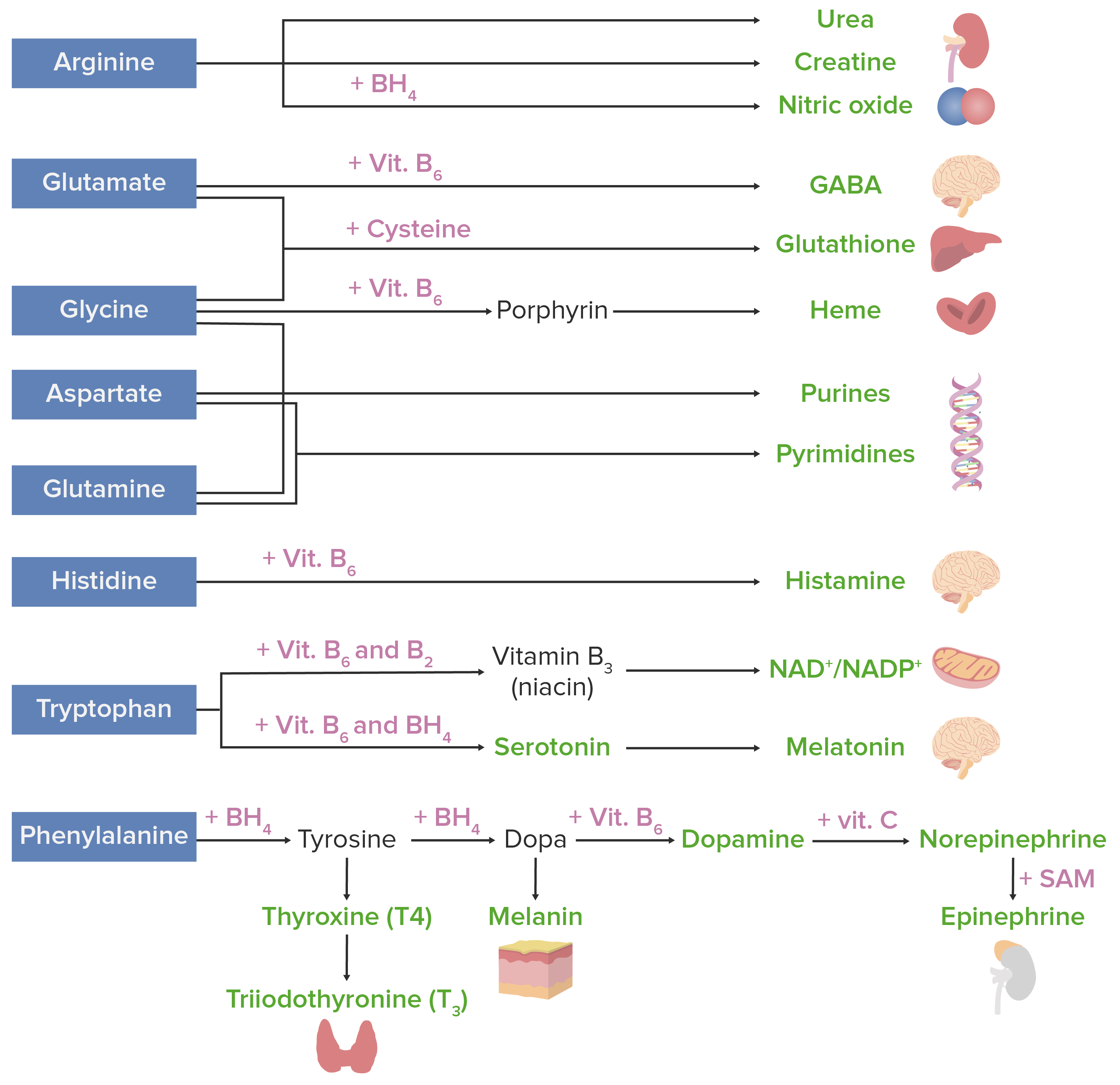
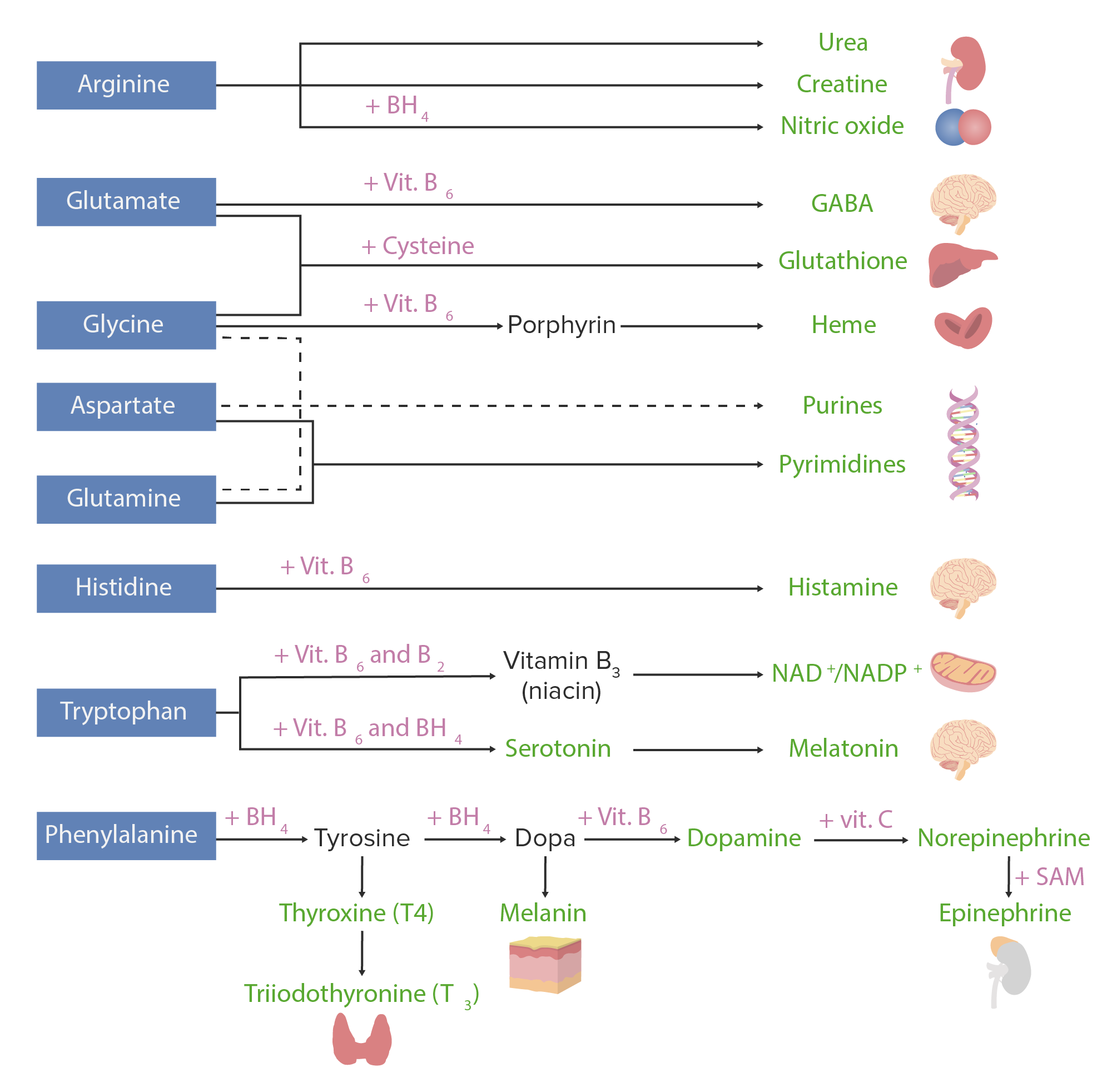
Amino acidsAmino acidsOrganic compounds that generally contain an amino (-NH2) and a carboxyl (-COOH) group. Twenty alpha-amino acids are the subunits which are polymerized to form proteins.Basics of Amino Acids can be used to assemble many substances. The image below shows the most important AA-derived substances in humans.
Amino acid derivatives. Amino acids (in blue) are combined with certain cofactors or other substrates (in pink) to make several biologically-important substances (in green).
Transamination is the transfer of an amino group from an alpha-AA to an alpha-keto acid, which is an AAAAAmyloidosis with an alpha-keto group (=O) instead of an alpha-amino group (NH2).
The original AAAAAmyloidosis loses an amino group and gains a keto group, becoming an alpha-keto acid.
The original alpha-keto acid loses its keto group and gains an amino, becoming a nonessential AAAAAmyloidosis.
The reaction is catalyzed by aminotransferase enzymesEnzymesEnzymes are complex protein biocatalysts that accelerate chemical reactions without being consumed by them. Due to the body’s constant metabolic needs, the absence of enzymes would make life unsustainable, as reactions would occur too slowly without these molecules. Basics of Enzymes:
Can be specific for a particular AAAAAmyloidosis pair or a group with similar chemical compositions
Requires coenzyme pyridoxal phosphatePhosphateInorganic salts of phosphoric acid.Electrolytes (PLP, the active form of vitamin B6Vitamin B6Vitamin B 6 refers to several picolines (especially pyridoxine; pyridoxal; & pyridoxamine) that are efficiently converted by the body to pyridoxal phosphate which is a coenzyme for synthesis of amino acids, neurotransmitters (serotonin, norepinephrine), sphingolipids, and aminolevulinic acid. During transamination of amino acids, pyridoxal phosphate is transiently converted into pyridoxamine phosphate. Although pyridoxine and vitamin B 6 are still frequently used as synonyms, especially by medical researchers, this practice is erroneous and sometimes misleading. Most of vitamin b6 is eventually degraded to pyridoxic acid and excreted in the urine.Water-soluble Vitamins and their Deficiencies)
Found in high concentrations in the liverLiverThe liver is the largest gland in the human body. The liver is found in the superior right quadrant of the abdomen and weighs approximately 1.5 kilograms. Its main functions are detoxification, metabolism, nutrient storage (e.g., iron and vitamins), synthesis of coagulation factors, formation of bile, filtration, and storage of blood. Liver: Anatomy
This process is need-dependent. If there is an excess of a type of AAAAAmyloidosis, the amino group of that type can be transferred to make other types of AAs that the body currently needs.
All of the common AAs participate in transamination, except lysine, threonine, prolineProlineA non-essential amino acid that is synthesized from glutamic acid. It is an essential component of collagen and is important for proper functioning of joints and tendons.Synthesis of Nonessential Amino Acids, and hydroxyproline, which catabolize via a dehydrogenase.
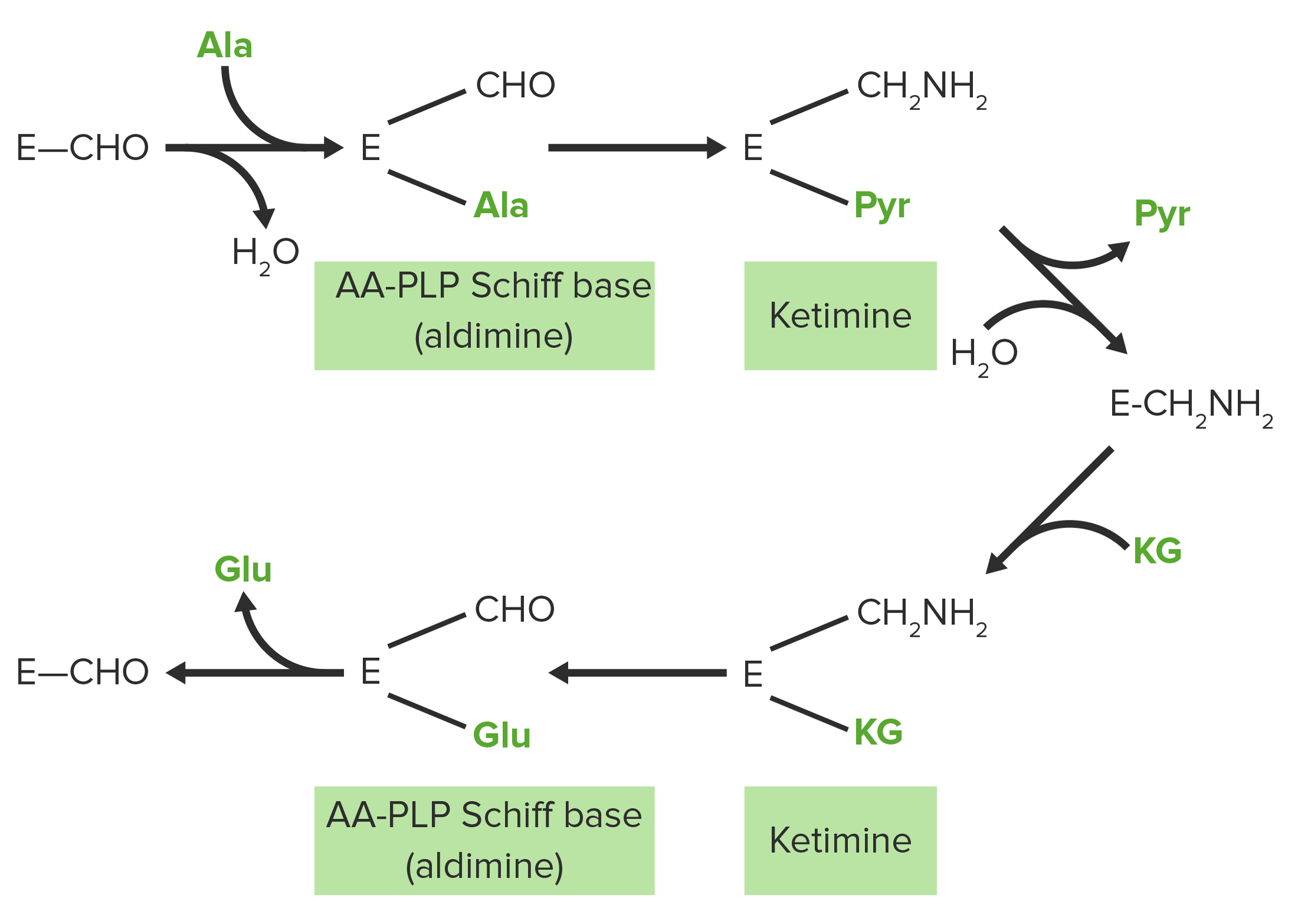
Schematic diagram of the transamination reactions of aspartate and glutamate (glutamic acid): Amino groups are highlighted in red. Keto groups are highlighted in green.
Image from Lecturio
TransaminasesTransaminasesA subclass of enzymes of the transferase class that catalyze the transfer of an amino group from a donor (generally an amino acid) to an acceptor (generally a 2-keto acid). Most of these enzymes are pyridoxyl phosphate proteins.Autoimmune Hepatitis
AlanineAlanineA non-essential amino acid that occurs in high levels in its free state in plasma. It is produced from pyruvate by transamination. It is involved in sugar and acid metabolism, increases immunity, and provides energy for muscle tissue, brain, and the central nervous system.Synthesis of Nonessential Amino Acids transaminase (ALTALTAn enzyme that catalyzes the conversion of l-alanine and 2-oxoglutarate to pyruvate and l-glutamate.Liver Function Tests or ALAT) transfers an amino group from alanineAlanineA non-essential amino acid that occurs in high levels in its free state in plasma. It is produced from pyruvate by transamination. It is involved in sugar and acid metabolism, increases immunity, and provides energy for muscle tissue, brain, and the central nervous system.Synthesis of Nonessential Amino Acids to alpha-ketoglutarate, forming pyruvatePyruvateDerivatives of pyruvic acid, including its salts and esters.Glycolysis and glutamateGlutamateDerivatives of glutamic acid. Included under this heading are a broad variety of acid forms, salts, esters, and amides that contain the 2-aminopentanedioic acid structure.Synthesis of Nonessential Amino Acids.
AspartateAspartateOne of the non-essential amino acids commonly occurring in the l-form. It is found in animals and plants, especially in sugar cane and sugar beets. It may be a neurotransmitter.Synthesis of Nonessential Amino Acids transaminase (ASTASTEnzymes of the transferase class that catalyze the conversion of l-aspartate and 2-ketoglutarate to oxaloacetate and l-glutamate.Liver Function Tests or ASAT)transfers an amino group from aspartateAspartateOne of the non-essential amino acids commonly occurring in the l-form. It is found in animals and plants, especially in sugar cane and sugar beets. It may be a neurotransmitter.Synthesis of Nonessential Amino Acids to alpha-ketoglutarate, forming oxaloacetateOxaloacetateDerivatives of oxaloacetic acid. Included under this heading are a broad variety of acid forms, salts, esters, and amides that include a 2-keto-1, 4-carboxy aliphatic structure.Citric Acid Cycle and glutamateGlutamateDerivatives of glutamic acid. Included under this heading are a broad variety of acid forms, salts, esters, and amides that contain the 2-aminopentanedioic acid structure.Synthesis of Nonessential Amino Acids.
Both enzymesEnzymesEnzymes are complex protein biocatalysts that accelerate chemical reactions without being consumed by them. Due to the body’s constant metabolic needs, the absence of enzymes would make life unsustainable, as reactions would occur too slowly without these molecules. Basics of Enzymes catabolize reversible reactions, which are essential for the transport of nitrogenNitrogenAn element with the atomic symbol n, atomic number 7, and atomic weight [14. 00643; 14. 00728]. Nitrogen exists as a diatomic gas and makes up about 78% of the earth’s atmosphere by volume. It is a constituent of proteins and nucleic acids and found in all living cells.Urea Cycle from tissues to the liverLiverThe liver is the largest gland in the human body. The liver is found in the superior right quadrant of the abdomen and weighs approximately 1.5 kilograms. Its main functions are detoxification, metabolism, nutrient storage (e.g., iron and vitamins), synthesis of coagulation factors, formation of bile, filtration, and storage of blood. Liver: Anatomy and into the ureaUreaA compound formed in the liver from ammonia produced by the deamination of amino acids. It is the principal end product of protein catabolism and constitutes about one half of the total urinary solids.Urea Cycle cycle.
Ping pong bi-bi mechanism of PLP-dependent enzyme-catalyzed transamination. Aminotransferase reaction occurs in 2 stages consisting of 3 steps: transamination, tautomerization, and hydrolysis. In the first stage, the alpha-amino group of the amino acid is transferred to PLP, yielding an alpha ketoacid and pyridoxamine phosphate (PMP). In the second stage of the reaction, the amino group of PMP is transferred to a different alpha-keto acid to yield a new alpha-amino acid and PLP.
Image by Lecturio.
Steps:
PLP reacts with the amino group of the AAAAAmyloidosis, releasing H2O.
A Schiff base is formed, destabilizing the AAAAAmyloidosis.
Hydrogen atoms migrate, double-bond shifts, and aldimine → ketimine.
H2O is added, yielding PMP and an alpha-keto acid.
In reverse, PMP reacts with an alpha-keto acid, generating an AAAAAmyloidosis and reconstituting PLP.
Deamination is the process through which amino groups are stripped from AAs, releasing free cytotoxicCytotoxicParvovirus B19ammoniaAmmoniaA colorless alkaline gas. It is formed in the body during decomposition of organic materials during a large number of metabolically important reactions. Note that the aqueous form of ammonia is referred to as ammonium hydroxide.Acid-Base Balance: ammoniaAmmoniaA colorless alkaline gas. It is formed in the body during decomposition of organic materials during a large number of metabolically important reactions. Note that the aqueous form of ammonia is referred to as ammonium hydroxide.Acid-Base Balance → ammonium → ureaUreaA compound formed in the liver from ammonia produced by the deamination of amino acids. It is the principal end product of protein catabolism and constitutes about one half of the total urinary solids.Urea Cycle or uric acidUric acidAn oxidation product, via xanthine oxidase, of oxypurines such as xanthine and hypoxanthine. It is the final oxidation product of purine catabolism in humans and primates, whereas in most other mammals urate oxidase further oxidizes it to allantoin.Nephrolithiasis via the ureaUreaA compound formed in the liver from ammonia produced by the deamination of amino acids. It is the principal end product of protein catabolism and constitutes about one half of the total urinary solids.Urea Cycle cycle in the liverLiverThe liver is the largest gland in the human body. The liver is found in the superior right quadrant of the abdomen and weighs approximately 1.5 kilograms. Its main functions are detoxification, metabolism, nutrient storage (e.g., iron and vitamins), synthesis of coagulation factors, formation of bile, filtration, and storage of blood. Liver: Anatomy.
Three types of deamination
1. Oxidative deamination:
Oxidation turns the amino group into an imino group.
NADNAD+A coenzyme composed of ribosylnicotinamide 5′-diphosphate coupled to adenosine 5′-phosphate by pyrophosphate linkage. It is found widely in nature and is involved in numerous enzymatic reactions in which it serves as an electron carrier by being alternately oxidized (NAD+) and reduced (NADH). Pentose Phosphate Pathway+ or NADP+ is reduced to NADH/H or NADPHNADPHNicotinamide adenine dinucleotide phosphate. A coenzyme composed of ribosylnicotinamide 5′-phosphate (nmn) coupled by pyrophosphate linkage to the 5′-phosphate adenosine 2.Pentose Phosphate Pathway/H, respectively.
Water is added to the amino group, converting it to an alpha-keto group, releasing ammoniaAmmoniaA colorless alkaline gas. It is formed in the body during decomposition of organic materials during a large number of metabolically important reactions. Note that the aqueous form of ammonia is referred to as ammonium hydroxide.Acid-Base Balance.
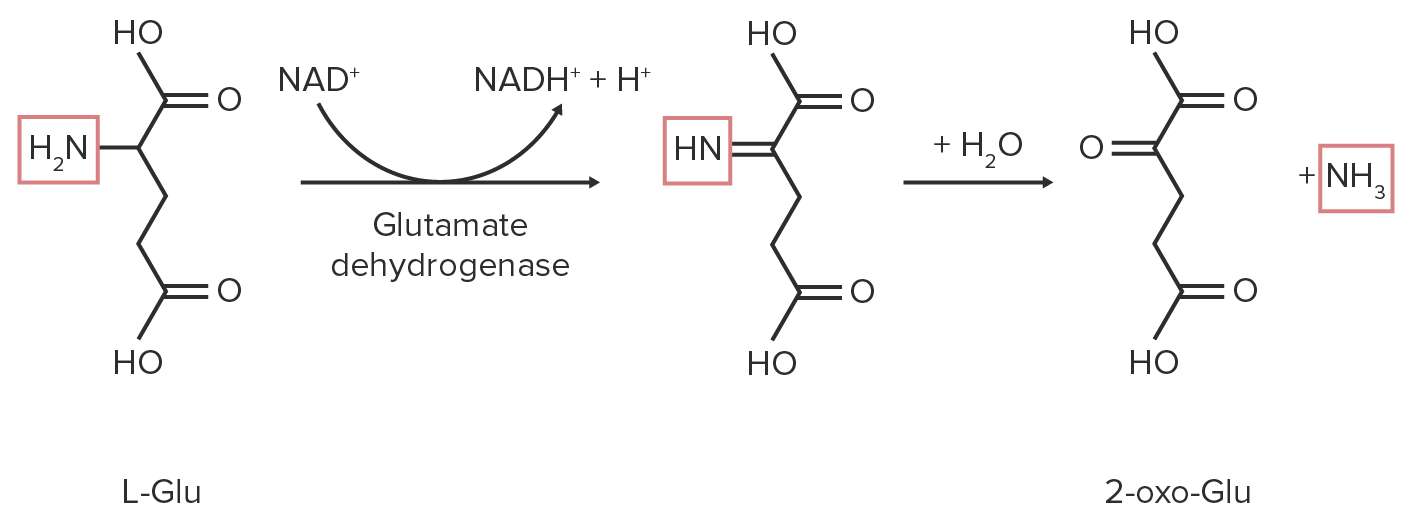
Schematic diagram of the oxidative deamination reaction of glutamate. The nitrogen-containing functional groups are highlighted in red.
Image by Lecturio.
2. Hydrolytic deamination:
Water reacts with the amino group, irreversibly attaching an OH group and eliminating the amino group in the form of ammoniaAmmoniaA colorless alkaline gas. It is formed in the body during decomposition of organic materials during a large number of metabolically important reactions. Note that the aqueous form of ammonia is referred to as ammonium hydroxide.Acid-Base Balance.
Schematic diagram of a hydrolytic deamination reaction. The nitrogen-containing functional groups are highlighted in red.
Image by Lecturio.
3. Eliminative deamination:
Small AAs (serineSerineA non-essential amino acid occurring in natural form as the l-isomer. It is synthesized from glycine or threonine. It is involved in the biosynthesis of purines; pyrimidines; and other amino acids.Synthesis of Nonessential Amino Acids or cysteineCysteineA thiol-containing non-essential amino acid that is oxidized to form cystine.Synthesis of Nonessential Amino Acids) release water (or hydrogen sulfideHydrogen sulfideA flammable, poisonous gas with a characteristic odor of rotten eggs. It is used in the manufacture of chemicals, in metallurgy, and as an analytical reagent.Salmonella for sulfurous amino acidsAmino acidsOrganic compounds that generally contain an amino (-NH2) and a carboxyl (-COOH) group. Twenty alpha-amino acids are the subunits which are polymerized to form proteins.Basics of Amino Acids).
PLP is a necessary coenzyme.
Through hydrolysisHydrolysisThe process of cleaving a chemical compound by the addition of a molecule of water.Proteins and Peptides, the amino group is cleaved, resulting in pyruvatePyruvateDerivatives of pyruvic acid, including its salts and esters.Glycolysis.
Schematic diagram of the eliminative deamination reaction of serine. The nitrogen-containing functional groups are highlighted in red, while the water molecule (H2O) and its components are highlighted in green.
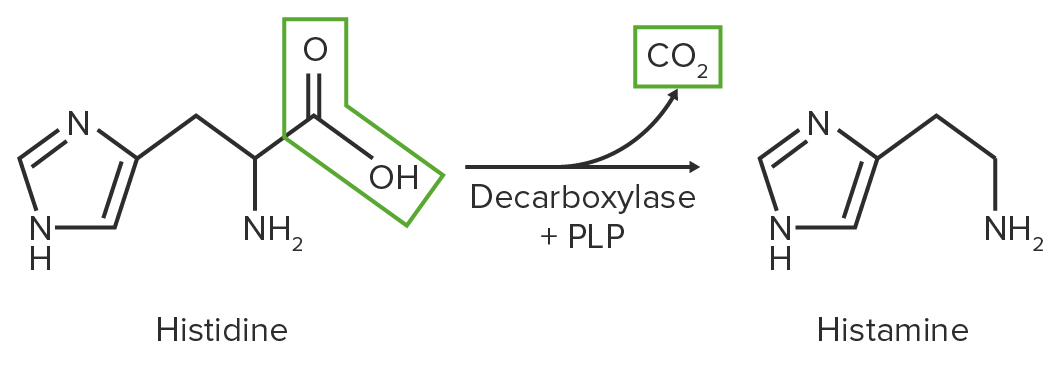
Cleavage of a carboxyl group from an AAAAAmyloidosis, releasing CO2
Catalyzed by the enzyme decarboxylase
Uses PLP as a coenzyme
Resulting amines fulfill important functions in the body = biogenic amines
Histamine is formed through decarboxylation from histidine, and plays a vital role in immediate hypersensitivity reactions.
Other examples:
Gamma-aminobutyric acid from glutamineGlutamineA non-essential amino acid present abundantly throughout the body and is involved in many metabolic processes. It is synthesized from glutamic acid and ammonia. It is the principal carrier of nitrogen in the body and is an important energy source for many cells.Synthesis of Nonessential Amino Acids acid
DopamineDopamineOne of the catecholamine neurotransmitters in the brain. It is derived from tyrosine and is the precursor to norepinephrine and epinephrine. Dopamine is a major transmitter in the extrapyramidal system of the brain, and important in regulating movement.Receptors and Neurotransmitters of the CNS from 3,4-dihydroxyphenylalanine
Schematic diagram of the decarboxylation reaction of histidine to histamine
Image by Lecturio.
Catabolism of the Carbon Skeleton
The catabolism of AAs involves anaplerotic reactions (chemical reactions that form intermediates of metabolic pathways).
The breakdown of the carbon skeleton of AAs can be classified by the metabolic pathways to which their catabolic products will serve as intermediates:
Glucogenic AAs → gluconeogenesisGluconeogenesisGluconeogenesis is the process of making glucose from noncarbohydrate precursors. This metabolic pathway is more than just a reversal of glycolysis. Gluconeogenesis provides the body with glucose not obtained from food, such as during a fasting period. The production of glucose is critical for organs and cells that cannot use fat for fuel. Gluconeogenesis intermediates
AlanineAlanineA non-essential amino acid that occurs in high levels in its free state in plasma. It is produced from pyruvate by transamination. It is involved in sugar and acid metabolism, increases immunity, and provides energy for muscle tissue, brain, and the central nervous system.Synthesis of Nonessential Amino Acids
ArginineArginineAn essential amino acid that is physiologically active in the l-form.Urea Cycle
AsparagineAsparagineA non-essential amino acid that is involved in the metabolic control of cell functions in nerve and brain tissue. It is biosynthesized from aspartic acid and ammonia by asparagine synthetase.Synthesis of Nonessential Amino Acids
Glutamic acidGlutamic acidA non-essential amino acid naturally occurring in the l-form. Glutamic acid is the most common excitatory neurotransmitter in the central nervous system.Urea Cycle
GlutamineGlutamineA non-essential amino acid present abundantly throughout the body and is involved in many metabolic processes. It is synthesized from glutamic acid and ammonia. It is the principal carrier of nitrogen in the body and is an important energy source for many cells.Synthesis of Nonessential Amino Acids
GlycineGlycineA non-essential amino acid. It is found primarily in gelatin and silk fibroin and used therapeutically as a nutrient. It is also a fast inhibitory neurotransmitter.Synthesis of Nonessential Amino Acids
Histidine
Methionine
ProlineProlineA non-essential amino acid that is synthesized from glutamic acid. It is an essential component of collagen and is important for proper functioning of joints and tendons.Synthesis of Nonessential Amino Acids
SerineSerineA non-essential amino acid occurring in natural form as the l-isomer. It is synthesized from glycine or threonine. It is involved in the biosynthesis of purines; pyrimidines; and other amino acids.Synthesis of Nonessential Amino Acids
TyrosineTyrosineA non-essential amino acid. In animals it is synthesized from phenylalanine. It is also the precursor of epinephrine; thyroid hormones; and melanin.Synthesis of Nonessential Amino Acids
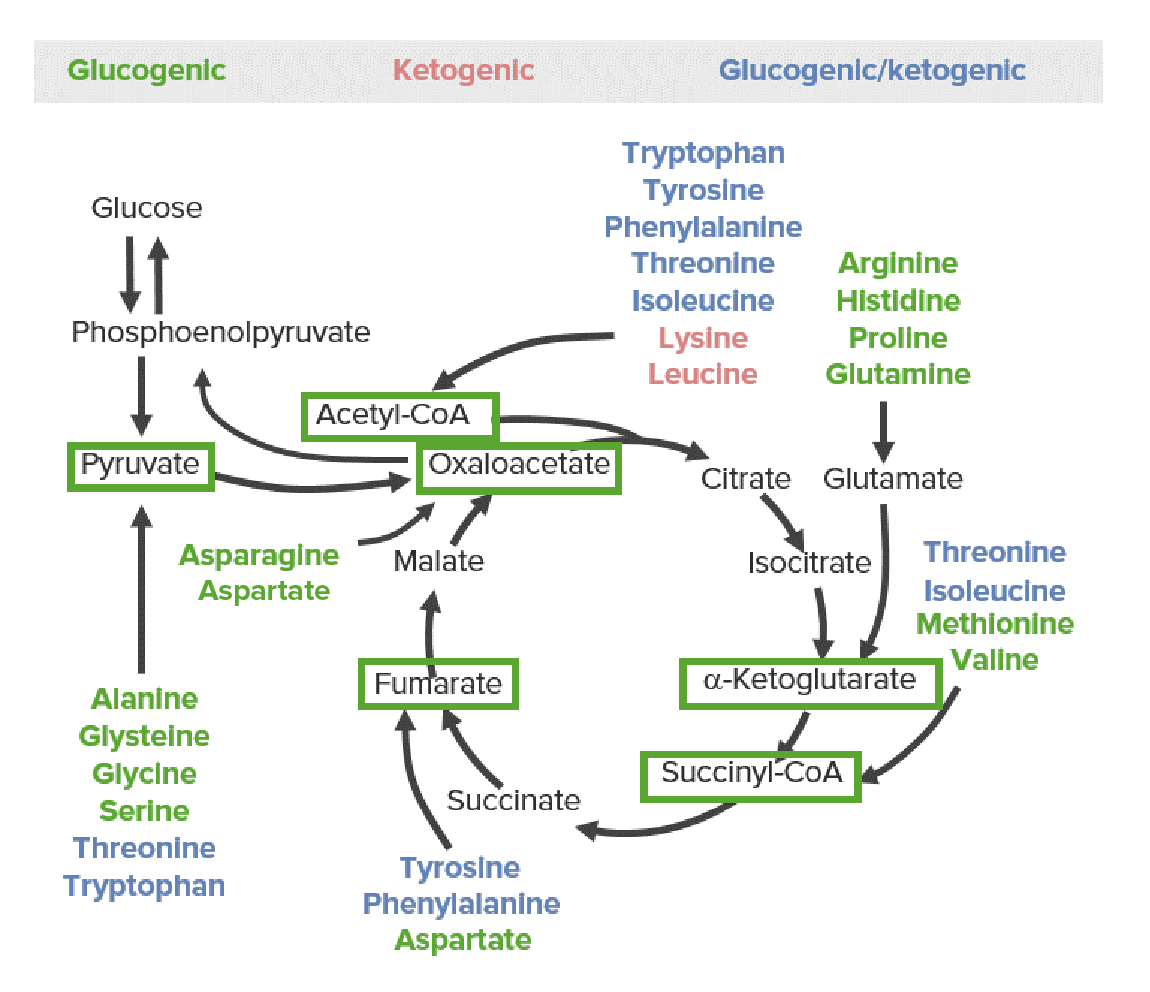
All AAs are broken down into 1 of 6 intermediates (see green boxes in the images below): pyruvatePyruvateDerivatives of pyruvic acid, including its salts and esters.Glycolysis, acetyl-CoAAcetyl-CoAAcetyl CoA participates in the biosynthesis of fatty acids and sterols, in the oxidation of fatty acids and in the metabolism of many amino acids. It also acts as a biological acetylating agent.Citric Acid Cycle, oxaloacetateOxaloacetateDerivatives of oxaloacetic acid. Included under this heading are a broad variety of acid forms, salts, esters, and amides that include a 2-keto-1, 4-carboxy aliphatic structure.Citric Acid Cycle, alpha-ketoglutarate, succinyl-CoASuccinyl-CoACitric Acid Cycle, or fumarateFumarateCitric Acid Cycle.
The 3 categories of catabolic products of amino acids: glucogenic (green), ketogenic (red), and both glucogenic and ketogenic (blue). The glucose-pyruvate pathway on the left represents glycolysis and gluconeogenesis. The cyclic pathway on the right represents the citric acid cycle. All amino acids are broken down into 1 of 6 intermediates (green boxes): pyruvate, acetyl-CoA, oxaloacetate, alpha-ketoglutarate, succinyl-CoA, or fumarate.
Glucogenic AAs
Metabolized to pyruvatePyruvateDerivatives of pyruvic acid, including its salts and esters.Glycolysis or metabolites of the citric acid cycleCitric acid cycleThe citric acid cycle, also known as the tricarboxylic acid (TCA) cycle or the Krebs cycle, is a cyclic set of reactions that occurs in the mitochondrial matrix. The TCA cycle is the continuation of any metabolic pathway that produces pyruvate, which is converted into its main substrate, acetyl-CoA. Citric Acid Cycle (CAC):
PyruvatePyruvateDerivatives of pyruvic acid, including its salts and esters.Glycolysis (from serineSerineA non-essential amino acid occurring in natural form as the l-isomer. It is synthesized from glycine or threonine. It is involved in the biosynthesis of purines; pyrimidines; and other amino acids.Synthesis of Nonessential Amino Acids, cysteineCysteineA thiol-containing non-essential amino acid that is oxidized to form cystine.Synthesis of Nonessential Amino Acids, glycineGlycineA non-essential amino acid. It is found primarily in gelatin and silk fibroin and used therapeutically as a nutrient. It is also a fast inhibitory neurotransmitter.Synthesis of Nonessential Amino Acids, alanineAlanineA non-essential amino acid that occurs in high levels in its free state in plasma. It is produced from pyruvate by transamination. It is involved in sugar and acid metabolism, increases immunity, and provides energy for muscle tissue, brain, and the central nervous system.Synthesis of Nonessential Amino Acids, and threonine)
OxaloacetateOxaloacetateDerivatives of oxaloacetic acid. Included under this heading are a broad variety of acid forms, salts, esters, and amides that include a 2-keto-1, 4-carboxy aliphatic structure.Citric Acid Cycle (from asparagineAsparagineA non-essential amino acid that is involved in the metabolic control of cell functions in nerve and brain tissue. It is biosynthesized from aspartic acid and ammonia by asparagine synthetase.Synthesis of Nonessential Amino Acids via aspartateAspartateOne of the non-essential amino acids commonly occurring in the l-form. It is found in animals and plants, especially in sugar cane and sugar beets. It may be a neurotransmitter.Synthesis of Nonessential Amino Acids)
α-Ketoglutarate (from glutamineGlutamineA non-essential amino acid present abundantly throughout the body and is involved in many metabolic processes. It is synthesized from glutamic acid and ammonia. It is the principal carrier of nitrogen in the body and is an important energy source for many cells.Synthesis of Nonessential Amino Acids, arginineArginineAn essential amino acid that is physiologically active in the l-form.Urea Cycle, histidine, prolineProlineA non-essential amino acid that is synthesized from glutamic acid. It is an essential component of collagen and is important for proper functioning of joints and tendons.Synthesis of Nonessential Amino Acids via glutamateGlutamateDerivatives of glutamic acid. Included under this heading are a broad variety of acid forms, salts, esters, and amides that contain the 2-aminopentanedioic acid structure.Synthesis of Nonessential Amino Acids)
Catabolic products either move into the CAC to produce energy or are used as substrates for gluconeogenesisGluconeogenesisGluconeogenesis is the process of making glucose from noncarbohydrate precursors. This metabolic pathway is more than just a reversal of glycolysis. Gluconeogenesis provides the body with glucose not obtained from food, such as during a fasting period. The production of glucose is critical for organs and cells that cannot use fat for fuel. Gluconeogenesis.
Ketogenic AAs
Metabolized directly to acetyl-CoAAcetyl-CoAAcetyl CoA participates in the biosynthesis of fatty acids and sterols, in the oxidation of fatty acids and in the metabolism of many amino acids. It also acts as a biological acetylating agent.Citric Acid Cycle, then enter 1 of 3 metabolic pathways:
Enter the CAC to produce ATP/energy
KetogenesisKetogenesisKetone Body Metabolism (production of ketone bodiesKetone bodiesThe metabolic substances acetone; 3-hydroxybutyric acid; and acetoacetic acid (acetoacetates). They are produced in the liver and kidney during fatty acids oxidation and used as a source of energy by the heart, muscle and brain.Ketone Body Metabolism)
SynthesisSynthesisPolymerase Chain Reaction (PCR) of fatty acidsAcidsChemical compounds which yield hydrogen ions or protons when dissolved in water, whose hydrogen can be replaced by metals or basic radicals, or which react with bases to form salts and water (neutralization). An extension of the term includes substances dissolved in media other than water.Acid-Base Balance or cholesterolCholesterolThe principal sterol of all higher animals, distributed in body tissues, especially the brain and spinal cord, and in animal fats and oils.Cholesterol Metabolism
Glucogenic and Ketogenic AAs
Metabolized to intermediates of lipidic as well as glucogenic pathways:
PhenylalaninePhenylalanineAn essential aromatic amino acid that is a precursor of melanin; dopamine; noradrenalin (norepinephrine), and thyroxine.Synthesis of Nonessential Amino Acids → tyrosineTyrosineA non-essential amino acid. In animals it is synthesized from phenylalanine. It is also the precursor of epinephrine; thyroid hormones; and melanin.Synthesis of Nonessential Amino Acids → fumarateFumarateCitric Acid Cycle and acetyl-CoAAcetyl-CoAAcetyl CoA participates in the biosynthesis of fatty acids and sterols, in the oxidation of fatty acids and in the metabolism of many amino acids. It also acts as a biological acetylating agent.Citric Acid Cycle
Threonine → propionyl-CoA and pyruvatePyruvateDerivatives of pyruvic acid, including its salts and esters.Glycolysis as well as acetyl-CoAAcetyl-CoAAcetyl CoA participates in the biosynthesis of fatty acids and sterols, in the oxidation of fatty acids and in the metabolism of many amino acids. It also acts as a biological acetylating agent.Citric Acid Cycle (via glycineGlycineA non-essential amino acid. It is found primarily in gelatin and silk fibroin and used therapeutically as a nutrient. It is also a fast inhibitory neurotransmitter.Synthesis of Nonessential Amino Acids + acetaldehydeAcetaldehydeA colorless, flammable liquid used in the manufacture of acetic acid, perfumes, and flavors. It is also an intermediate in the metabolism of alcohol. It has a general narcotic action and also causes irritation of mucous membranes. Large doses may cause death from respiratory paralysis.Ethanol Metabolism)
Tryptophan → alanineAlanineA non-essential amino acid that occurs in high levels in its free state in plasma. It is produced from pyruvate by transamination. It is involved in sugar and acid metabolism, increases immunity, and provides energy for muscle tissue, brain, and the central nervous system.Synthesis of Nonessential Amino Acids and acetyl-CoAAcetyl-CoAAcetyl CoA participates in the biosynthesis of fatty acids and sterols, in the oxidation of fatty acids and in the metabolism of many amino acids. It also acts as a biological acetylating agent.Citric Acid Cycle
Mnemonic
To recall the metabolic pathways of the carbon skeletons of amino acidsAmino acidsOrganic compounds that generally contain an amino (-NH2) and a carboxyl (-COOH) group. Twenty alpha-amino acids are the subunits which are polymerized to form proteins.Basics of Amino Acids, remember:
Ketogenic: “The onLy pureLy ketogenic amino acidsAmino acidsOrganic compounds that generally contain an amino (-NH2) and a carboxyl (-COOH) group. Twenty alpha-amino acids are the subunits which are polymerized to form proteins.Basics of Amino Acids.”
Leucine
Lysine
Clinical Relevance
The following conditions are disorders of amino acid metabolismDisorders of amino acid metabolismAmino acids are the building blocks of proteins, whose production is a tightly regulated enzymatic cascade. Disorders of the enzymes involved in amino acid metabolism are often serious and present early in life. Congenital errors in amino acid metabolism are due to either impaired synthesis or degradation.Disorders of Amino Acid Metabolism. Depending on the country and the individual U.S. state, newbornNewbornAn infant during the first 28 days after birth.Physical Examination of the Newborn infants may be routinely screened for these disorders (except for alkaptonuria).
Phenylketonuria: a defect of phenylalanine hydroxylasePhenylalanine hydroxylaseAn enzyme of the oxidoreductase class that catalyzes the formation of l-tyrosine, dihydrobiopterin, and water from l-phenylalanine, tetrahydrobiopterin, and oxygen.Synthesis of Nonessential Amino Acids that results in the impairment of the conversion of phenylalaninePhenylalanineAn essential aromatic amino acid that is a precursor of melanin; dopamine; noradrenalin (norepinephrine), and thyroxine.Synthesis of Nonessential Amino Acids to tyrosineTyrosineA non-essential amino acid. In animals it is synthesized from phenylalanine. It is also the precursor of epinephrine; thyroid hormones; and melanin.Synthesis of Nonessential Amino Acids and subsequent accumulation of phenylalaninePhenylalanineAn essential aromatic amino acid that is a precursor of melanin; dopamine; noradrenalin (norepinephrine), and thyroxine.Synthesis of Nonessential Amino Acids. Presents as psychomotor delay and seizuresSeizuresA seizure is abnormal electrical activity of the neurons in the cerebral cortex that can manifest in numerous ways depending on the region of the brain affected. Seizures consist of a sudden imbalance that occurs between the excitatory and inhibitory signals in cortical neurons, creating a net excitation. The 2 major classes of seizures are focal and generalized. Seizures
Maple syrup urine diseaseMaple syrup urine diseaseAn autosomal recessive inherited disorder with multiple forms of phenotypic expression, caused by a defect in the oxidative decarboxylation of branched-chain amino acids. These metabolites accumulate in body fluids and render a ‘maple syrup’ odor. The disease is divided into classic, intermediate, intermittent, and thiamine responsive subtypes. The classic form presents in the first week of life with ketoacidosis, hypoglycemia, emesis, neonatal seizures, and hypertonia. The intermediate and intermittent forms present in childhood or later with acute episodes of ataxia and vomiting.Disorders of Amino Acid Metabolism: a defect in dehydrogenase that results in the accumulation of branched-chain AAs. Presents as cognitive disabilityDisabilityDetermination of the degree of a physical, mental, or emotional handicap. The diagnosis is applied to legal qualification for benefits and income under disability insurance and to eligibility for social security and workman’s compensation benefits.ABCDE Assessment, sweet-smelling urine, and dystoniaDystoniaDystonia is a hyperkinetic movement disorder characterized by the involuntary contraction of muscles, resulting in abnormal postures or twisting and repetitive movements. Dystonia can present in various ways as may affect many different skeletal muscle groups. Dystonia
Homocystinuria: a defect in the enzyme cystathionine β-synthase, which leads to an accumulation of homocysteine. Presents as flushing, developmental delay, lensLensA transparent, biconvex structure of the eye, enclosed in a capsule and situated behind the iris and in front of the vitreous humor (vitreous body). It is slightly overlapped at its margin by the ciliary processes. Adaptation by the ciliary body is crucial for ocular accommodation.Eye: Anatomy dislocation, vascular disease, and osteoporosisOsteoporosisOsteoporosis refers to a decrease in bone mass and density leading to an increased number of fractures. There are 2 forms of osteoporosis: primary, which is commonly postmenopausal or senile; and secondary, which is a manifestation of immobilization, underlying medical disorders, or long-term use of certain medications. Osteoporosis
TyrosinemiaTyrosinemiaA group of disorders which have in common elevations of tyrosine in the blood and urine secondary to an enzyme deficiency. Type I tyrosinemia features episodic weakness, self-mutilation, hepatic necrosis, renal tubular injury, and seizures and is caused by a deficiency of the enzyme fumarylacetoacetase. Type II tyrosinemia features intellectual disability, painful corneal ulcers, and keratosis of the palms and plantar surfaces and is caused by a deficiency of the enzyme tyrosine transaminase. Type III tyrosinemia features intellectual disability and is caused by a deficiency of the enzyme 4-hydroxyphenylpyruvate dioxygenase.Renal Tubular Acidosis: a deficiency of fumarylacetoacetate hydrolase, the last enzyme in the tyrosineTyrosineA non-essential amino acid. In animals it is synthesized from phenylalanine. It is also the precursor of epinephrine; thyroid hormones; and melanin.Synthesis of Nonessential Amino Acids catabolism. Presents as liverLiverThe liver is the largest gland in the human body. The liver is found in the superior right quadrant of the abdomen and weighs approximately 1.5 kilograms. Its main functions are detoxification, metabolism, nutrient storage (e.g., iron and vitamins), synthesis of coagulation factors, formation of bile, filtration, and storage of blood. Liver: Anatomy disease, deficient weight gain, peripheral nerve disease, and kidney defects
Alkaptonuria: a deficiency of homogentisic acid dioxygenase, which impairs the normal degradation of tyrosineTyrosineA non-essential amino acid. In animals it is synthesized from phenylalanine. It is also the precursor of epinephrine; thyroid hormones; and melanin.Synthesis of Nonessential Amino Acids to fumarateFumarateCitric Acid Cycle. Presents as a bluish-black discoloration of connective tissues, arthritisArthritisAcute or chronic inflammation of joints.Osteoarthritis, and calcifications of various tissues.
Guoyao, W. (2022). Amino acids: Biochemistry and nutrition (2nd ed.). CRC Press.
Jiang, H., Wang, Y., & Li, M. (2025). Role of branched-chain amino acid catabolism in the regulation of adipocyte metabolism. Endocrinology, bqaf089. https://doi.org/10.1210/endocr/bqaf089
Knol, M. G. E., Verhave, J. C., & Navis, G. (2024). Amino acid metabolism in kidney health and disease. Nature Reviews Nephrology, 20(12), 771–788. https://doi.org/10.1038/s41581-024-00872-8
Ling, Z.-N., Jiang, Y.-F., Ru, J.-N., Lu, J.-H., & Ding, B. (2023). Amino acid metabolism in health and disease. Signal Transduction and Targeted Therapy, 8, 345. https://doi.org/10.1038/s41392-023-01569-3
Nelson, D. L., & Cox, M. M. (2021). Lehninger principles of biochemistry (8th ed.). W. H. Freeman and Company.
Rodwell, V. W., Bender, D. A., Botham, K. M., Kennelly, P. J., & Weil, P. A. (2022). Harper’s illustrated biochemistry (32nd ed.). McGraw-Hill Education.
Torres, N., Tovar, A. R., & Torres, L. F. (2023). Amino acid catabolism: An overlooked area of metabolism. Nutrients, 15(15), 3378. https://doi.org/10.3390/nu15153378
Create your free account or log in to continue reading!