Amino acids Amino acids Organic compounds that generally contain an amino (-NH2) and a carboxyl (-COOH) group. Twenty alpha-amino acids are the subunits which are polymerized to form proteins. Basics of Amino Acids are the building blocks of proteins Proteins Linear polypeptides that are synthesized on ribosomes and may be further modified, crosslinked, cleaved, or assembled into complex proteins with several subunits. The specific sequence of amino acids determines the shape the polypeptide will take, during protein folding, and the function of the protein. Energy Homeostasis, whose production is a tightly regulated enzymatic cascade. Disorders of the enzymes Enzymes Enzymes are complex protein biocatalysts that accelerate chemical reactions without being consumed by them. Due to the body's constant metabolic needs, the absence of enzymes would make life unsustainable, as reactions would occur too slowly without these molecules. Basics of Enzymes involved in amino acid Amino acid Amino acids (AAs) are composed of a central carbon atom attached to a carboxyl group, an amino group, a hydrogen atom, and a side chain (R group). Basics of Amino Acids metabolism are often serious and present early in life. Congenital errors in amino acid Amino acid Amino acids (AAs) are composed of a central carbon atom attached to a carboxyl group, an amino group, a hydrogen atom, and a side chain (R group). Basics of Amino Acids metabolism are due to either impaired synthesis Synthesis Polymerase Chain Reaction (PCR) or degradation. In the United States, neonates are routinely screened at birth for common disorders of amino acid Amino acid Amino acids (AAs) are composed of a central carbon atom attached to a carboxyl group, an amino group, a hydrogen atom, and a side chain (R group). Basics of Amino Acids metabolism, which include phenylketonuria (PKU), maple syrup urine disease (MSUD), homocystinuria, tyrosinemia Tyrosinemia A group of disorders which have in common elevations of tyrosine in the blood and urine secondary to an enzyme deficiency. Type I tyrosinemia features episodic weakness, self-mutilation, hepatic necrosis, renal tubular injury, and seizures and is caused by a deficiency of the enzyme fumarylacetoacetase. Type II tyrosinemia features intellectual disability, painful corneal ulcers, and keratosis of the palms and plantar surfaces and is caused by a deficiency of the enzyme tyrosine transaminase. Type III tyrosinemia features intellectual disability and is caused by a deficiency of the enzyme 4-hydroxyphenylpyruvate dioxygenase. Renal Tubular Acidosis, and alkaptonuria. Symptoms of these conditions often present early in life. Although treatment varies, most of these conditions require dietary changes, and some require protein supplementation or other medication regimens.
Last updated: May 19, 2025
Phenylketonuria (PKU) is an inherited disease that causes an increase in phenylalanine Phenylalanine An essential aromatic amino acid that is a precursor of melanin; dopamine; noradrenalin (norepinephrine), and thyroxine. Synthesis of Nonessential Amino Acids levels in the body due to the inability to metabolize this amino acid Amino acid Amino acids (AAs) are composed of a central carbon atom attached to a carboxyl group, an amino group, a hydrogen atom, and a side chain (R group). Basics of Amino Acids.
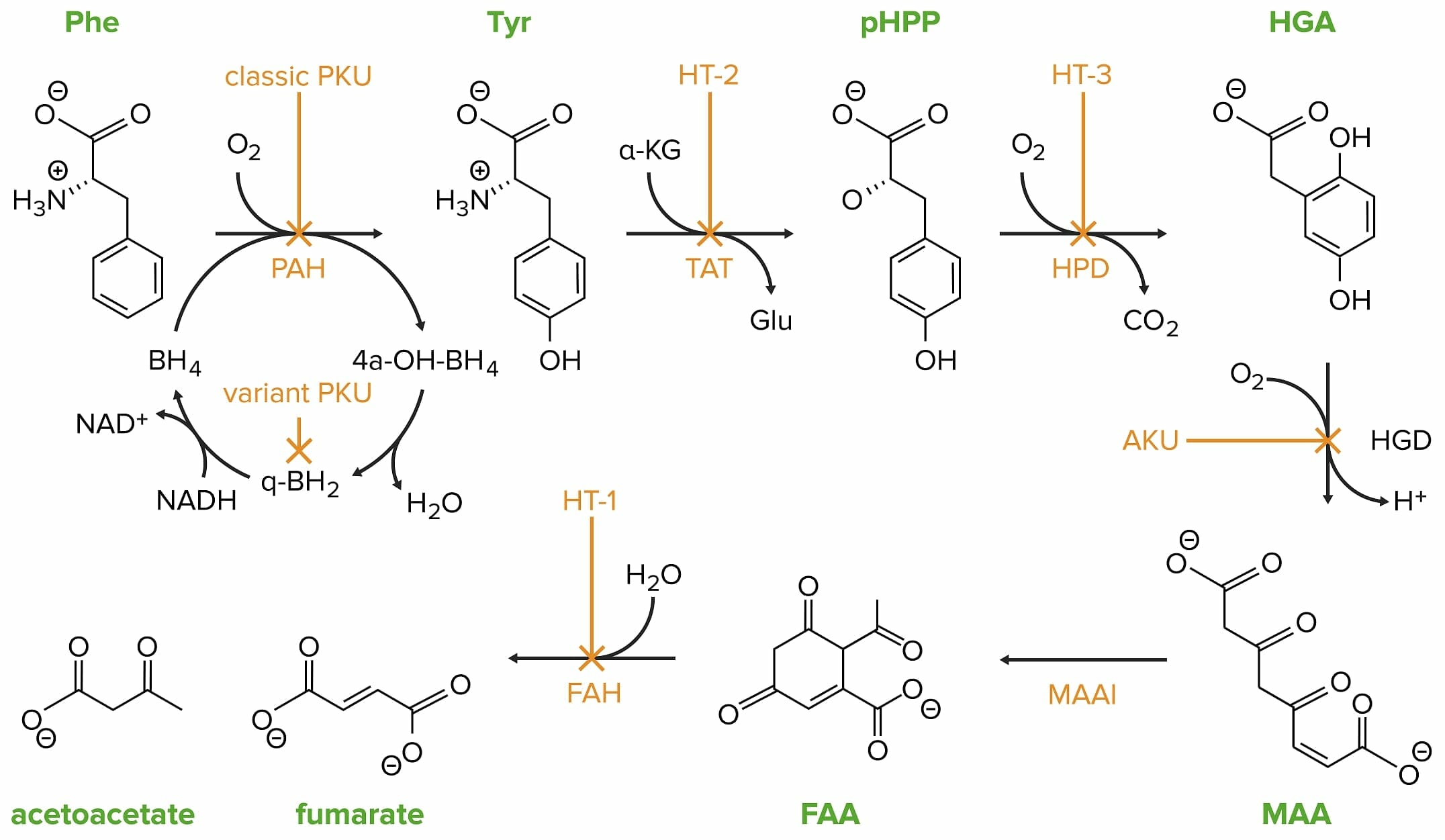
Classic phenylketonuria (PKU) is caused by a mutation in the phenylalanine hydroxylase (PAH) gene.
Phe: phenylalanine
PAH: phenylalanine hydroxylase
Tyr: tyrosine
HT-2: tyrosinemia II
TAT: tyrosine transaminase
pHPP: p-hydroxyphenylpyruvate
HPD: p-hydroxyphenylpyruvate dioxygenase
HT-3: tyrosinemia III
HGA: homogentisate
HGD: homogentisate oxidase
AKU: alcaptonuria
MAA: 4-maleylacetoacetate
MAAI: 4-maleylacetoacetate isomerase
FAA: 4-fumarylacetoacetate
FAH: 4-fumarylacetoacetase
HT-1: tyrosinemia I
The blood of a 2-week-old infant is collected for a phenylketonuria (PKU) screening.
Image: “Phenylketonuria testing” by USAF Photographic Archives. License: Public DomainThis condition cannot be cured and must be managed with dietary modification and medication/supplementation.
Cystinuria Cystinuria An inherited disorder due to defective reabsorption of cystine and other basic amino acids by the proximal renal tubules. This form of aminoaciduria is characterized by the abnormally high urinary levels of cystine; lysine; arginine; and ornithine. Mutations involve the amino acid transport protein gene slc3a1. Nephrolithiasis is a genetic cause of kidney stones Kidney stones Nephrolithiasis is the formation of a stone, or calculus, anywhere along the urinary tract caused by precipitations of solutes in the urine. The most common type of kidney stone is the calcium oxalate stone, but other types include calcium phosphate, struvite (ammonium magnesium phosphate), uric acid, and cystine stones. Nephrolithiasis due to impairment of renal cystine Cystine A covalently linked dimeric nonessential amino acid formed by the oxidation of cysteine. Two molecules of cysteine are joined together by a disulfide bridge to form cystine. Nephrolithiasis transport. There is decreased proximal tubular reabsorption of filtered cystine Cystine A covalently linked dimeric nonessential amino acid formed by the oxidation of cysteine. Two molecules of cysteine are joined together by a disulfide bridge to form cystine. Nephrolithiasis resulting in increased urinary cystine Cystine A covalently linked dimeric nonessential amino acid formed by the oxidation of cysteine. Two molecules of cysteine are joined together by a disulfide bridge to form cystine. Nephrolithiasis excretion and cystine Cystine A covalently linked dimeric nonessential amino acid formed by the oxidation of cysteine. Two molecules of cysteine are joined together by a disulfide bridge to form cystine. Nephrolithiasis stone formation.
Clinical suspicion:
Confirmatory testing:
Homocystinuria is an inherited metabolic disorder of methionine metabolism that results in elevated levels of homocysteine in the plasma Plasma The residual portion of blood that is left after removal of blood cells by centrifugation without prior blood coagulation. Transfusion Products and urine and causes an array of clinical manifestations.
There are 2 normal homocysteine metabolic pathways:
Elevations in plasma Plasma The residual portion of blood that is left after removal of blood cells by centrifugation without prior blood coagulation. Transfusion Products homocysteine levels primarily result from:
Other causes of elevated plasma Plasma The residual portion of blood that is left after removal of blood cells by centrifugation without prior blood coagulation. Transfusion Products homocysteine:
Clinical suspicion:
Confirmatory testing:
A genetic disorder manifesting in the abnormal metabolism of tyrosine Tyrosine A non-essential amino acid. In animals it is synthesized from phenylalanine. It is also the precursor of epinephrine; thyroid hormones; and melanin. Synthesis of Nonessential Amino Acids, an amino acid Amino acid Amino acids (AAs) are composed of a central carbon atom attached to a carboxyl group, an amino group, a hydrogen atom, and a side chain (R group). Basics of Amino Acids involved in the synthesis Synthesis Polymerase Chain Reaction (PCR) of thyroid Thyroid The thyroid gland is one of the largest endocrine glands in the human body. The thyroid gland is a highly vascular, brownish-red gland located in the visceral compartment of the anterior region of the neck. Thyroid Gland: Anatomy hormones Hormones Hormones are messenger molecules that are synthesized in one part of the body and move through the bloodstream to exert specific regulatory effects on another part of the body. Hormones play critical roles in coordinating cellular activities throughout the body in response to the constant changes in both the internal and external environments. Hormones: Overview and Types, catecholamines Catecholamines A general class of ortho-dihydroxyphenylalkylamines derived from tyrosine. Adrenal Hormones, and melanin Melanin Insoluble polymers of tyrosine derivatives found in and causing darkness in skin (skin pigmentation), hair, and feathers providing protection against sunburn induced by sunlight. Carotenes contribute yellow and red coloration. Seborrheic Keratosis.
Under nonpathological conditions, tyrosine Tyrosine A non-essential amino acid. In animals it is synthesized from phenylalanine. It is also the precursor of epinephrine; thyroid hormones; and melanin. Synthesis of Nonessential Amino Acids is metabolized by hepatocytes Hepatocytes The main structural component of the liver. They are specialized epithelial cells that are organized into interconnected plates called lobules. Liver: Anatomy and renal proximal tubules into acetoacetate Acetoacetate Salts and derivatives of acetoacetic acid. Ketone Body Metabolism (ketogenic) and fumarate Fumarate Citric Acid Cycle (glucogenic).
Genetic defects:
HT1:
HT2:
HT3:
AKU:
Clinical suspicion:
Confirmatory testing:
A group of genetic disorders of amino acid Amino acid Amino acids (AAs) are composed of a central carbon atom attached to a carboxyl group, an amino group, a hydrogen atom, and a side chain (R group). Basics of Amino Acids metabolism involving branched-chain amino acids Amino acids Organic compounds that generally contain an amino (-NH2) and a carboxyl (-COOH) group. Twenty alpha-amino acids are the subunits which are polymerized to form proteins. Basics of Amino Acids such as leucine, isoleucine, and valine. These disorders are caused by disruption of metabolism of these amino acids Amino acids Organic compounds that generally contain an amino (-NH2) and a carboxyl (-COOH) group. Twenty alpha-amino acids are the subunits which are polymerized to form proteins. Basics of Amino Acids, resulting in accumulation of toxic metabolites that often spill into urine.
There are 4 main types of organic acidemia Acidemia Respiratory Acidosis:
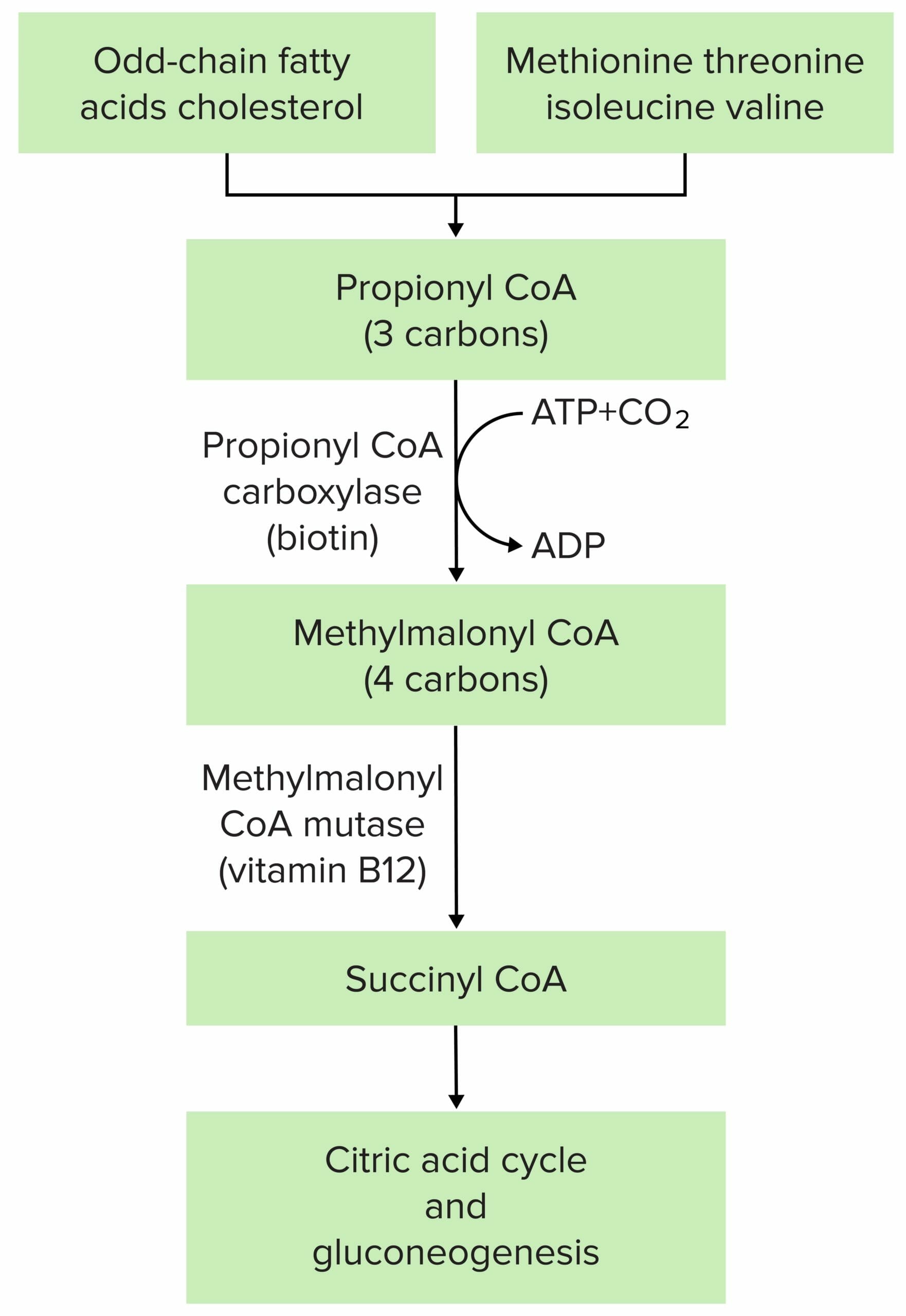
The pathophysiology of methylmalonic acidemia is most often due to mutation at either methylmalonyl CoA mutase or methylmalonyl CoA epimerase.
Image by Lecturio.