LosLOSNeisseria aminoácidos son losLOSNeisseria componentes básicos de las proteínas, cuya producción es una cascada enzimática estrechamente regulada. LosLOSNeisseria trastornos de las enzimas que intervienen enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el metabolismo de losLOSNeisseria aminoácidos son a menudo graves y se presentan enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum las primeras etapas de la vida. LosLOSNeisseria errores congénitos del metabolismo de losLOSNeisseria aminoácidos se deben a una alteración de la síntesis o de la degradación. EnENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema NodosumlosLOSNeisseria Estados Unidos, losLOSNeisseria neonatos son examinados rutinariamente alALAmyloidosis nacer para detectar trastornos comunes del metabolismo de losLOSNeisseria aminoácidos, que incluyen la fenilcetonuria, enfermedad de la orina con olor a jarabe de arce, homocistinuria, tirosinemia y alcaptonuria. LosLOSNeisseria síntomas de estas afecciones suelen presentarse enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum las primeras etapas de la vida. Aunque el tratamiento varía, la mayoría de estas afecciones requieren cambios enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la dieta, y algunas requieren suplementos de proteínas u otros regímenes de medicación.
La fenilcetonuria es una enfermedad hereditaria que provoca un aumento de losLOSNeisseria niveles de fenilalanina enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el organismo debido a la incapacidad de metabolizar este aminoácido.
Fisiopatología
Mutaciones enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el gen que codifica la fenilalanina hidroxilasa, una enzima que convierte la fenilalanina enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum tirosina
Un defecto enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la fenilalanina hidroxilasa conduce a la acumulación de fenilalanina (enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la mayoría de losLOSNeisseria casos losLOSNeisseria niveles de tirosina son normales o ligeramente bajos).
LosLOSNeisseria niveles elevados de fenilalanina causan daños enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema NodosumlosLOSNeisseria tractos de materia blanca y enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la mielina (mecanismo desconocido), lo que provoca déficits neurológicos.
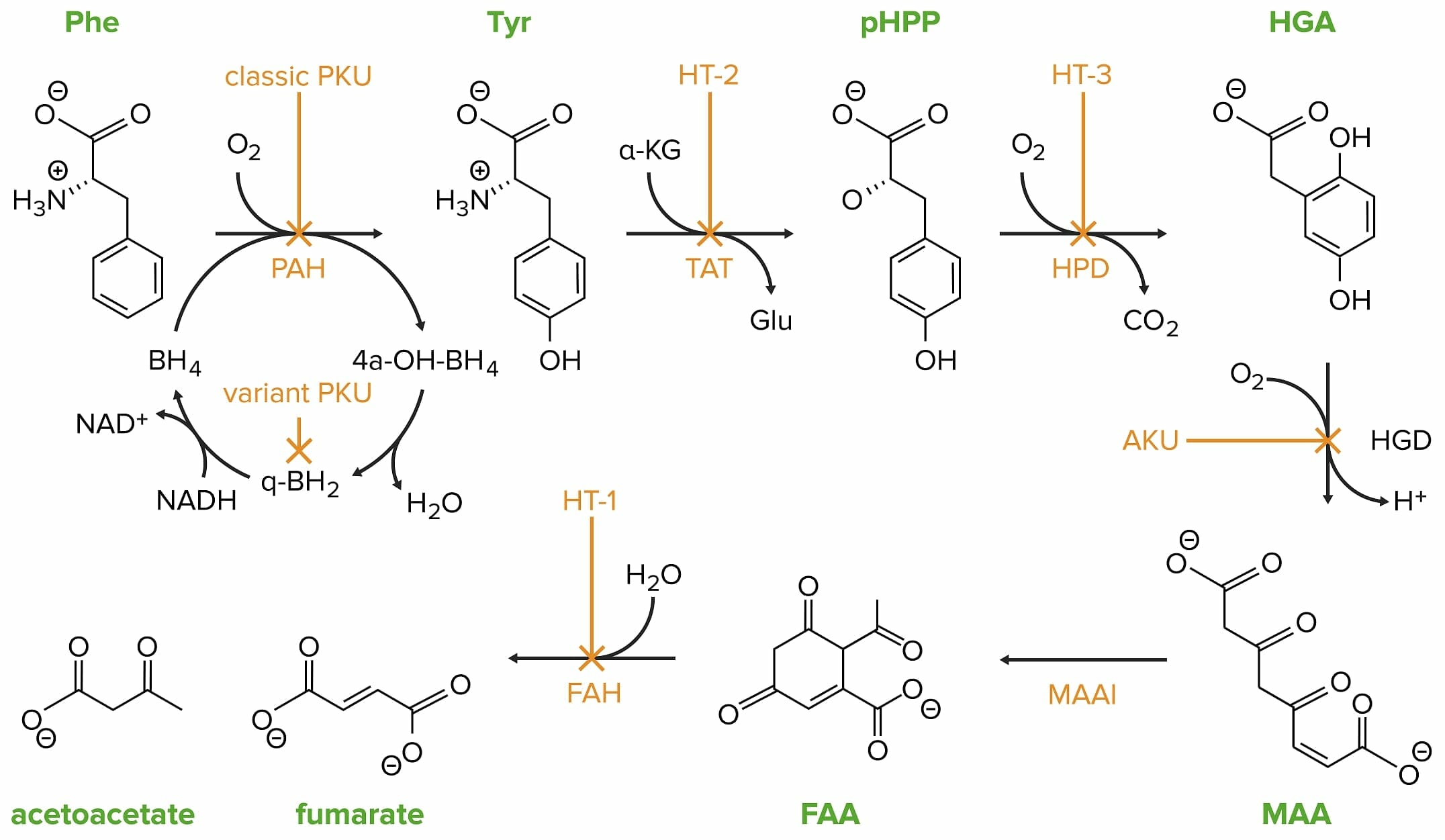
La fenilcetonuria clásica está causada por una mutación en el gen de la fenilalanina hidroxilasa. Phe: fenilalanina PAH: fenilalanina hidroxilasa Tyr: tirosina HT-2: tirosinemia II TAT: tirosina transaminasa pHPP: p-hidroxifenilpiruvato HPD: p-hidroxifenilpiruvato dioxigenasa HT-3: tirosinemia III HGA: homogentisato HGD: homogentisato oxidasa AKU: alcaptonuria MAA: 4-maleilacetoacetato MAAI: 4-maleilacetoacetato isomerasa FAA: 4-fumarilacetoacetato FAH: 4-fumarilacetoacetasa HT-1: tirosinemia I
Imagen por Lecturio.
Clasificación
Fenilcetonuria clásica: muy poca actividad del gen fenilalanina hidroxilasa y acumulación significativa de fenilalanina
Fenilcetonuria leve: acumulación de fenilalanina menos severa, cierta actividad del gen de la fenilalanina hidroxilasa presente
Síntomas
LosLOSNeisseria recién nacidos no presentan síntomas dado que la fenilalanina tarda enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum acumularse.
Neurológicos:
Retrasos enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el desarrollo
Discapacidad intelectual
Convulsiones
Microcefalia
Conductuales y psiquiátricos:
Problemas sociales
Problemas de comportamiento y emocionales
Problemas de salud mental
Hiperactividad
Ortopédicos: baja densidad ósea
Dermatológicos
Erupciones cutáneas (eczemaEczemaAtopic dermatitis, also known as eczema, is a chronic, relapsing, pruritic, inflammatory skin disease that occurs more frequently in children, although adults can also be affected. The condition is often associated with elevated serum levels of IgE and a personal or family history of atopy. Skin dryness, erythema, oozing, crusting, and lichenification are present. Atopic Dermatitis (Eczema))
Piel clara (por falta de melanina)
Otros: olor a humedad enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el aliento o enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la orina
Diagnóstico
Tamizaje de la fenilcetonuria mediante punción del talón enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la 1ra semana de vida
Medición de losLOSNeisseria niveles de aminoácidos que muestran una fenilalanina elevada y una tirosina baja/normal
LosLOSNeisseria resultados positivos deben confirmarse con pruebas de seguimiento.
Pruebas genéticas
Pruebas de portador enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema NodosumlosLOSNeisseria padres para el asesoramiento prenatal
Pruebas prenatales
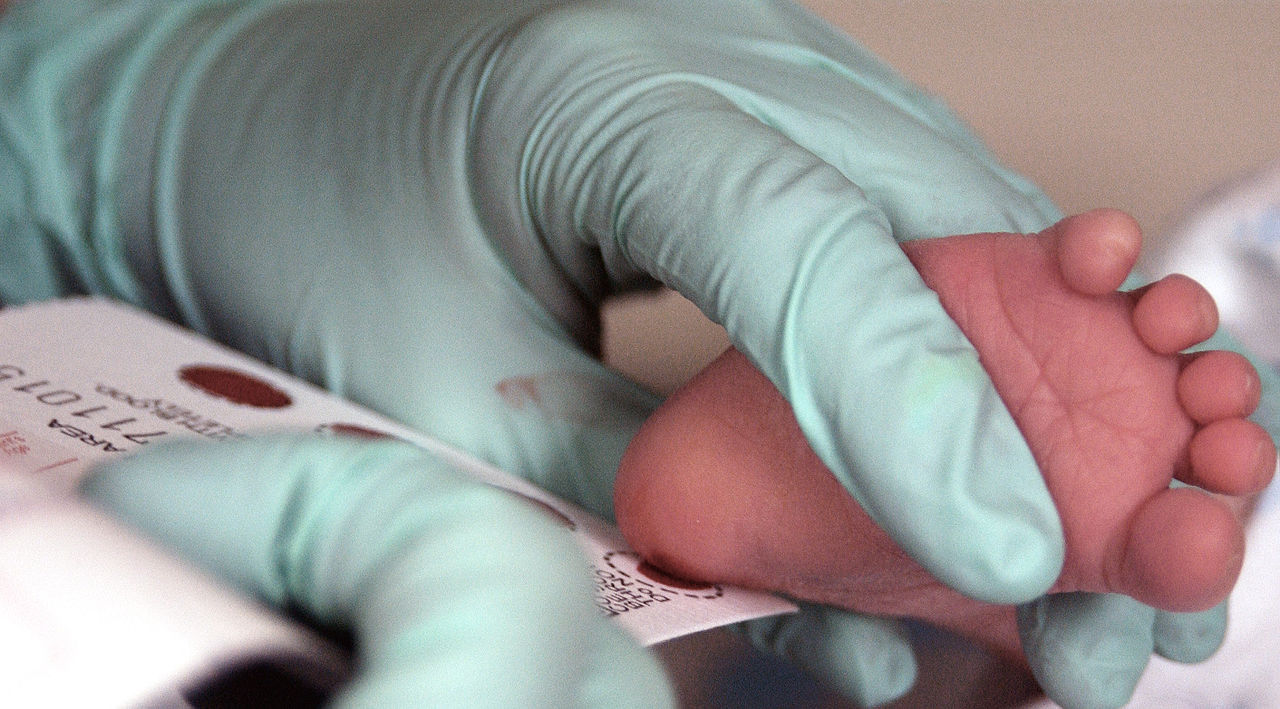
Se extrae sangre de un lactante de 2 semanas para realizar un tamizaje de fenilcetonuria.
Imagen: “Phenylketonuria testing” por USAF Photographic Archives. Licencia: Dominio Público
Tratamiento
Esta afección no puede curarse y debe tratarse con modificaciones enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la dieta y medicación/suplementos.
Análisis de sangre regulares para evaluar el nivel de fenilalanina enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la sangre
Dieta baja enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum fenilalanina, eliminando alimentos como soya, pollo, camarones, frutos secos, pavo y legumbres
Evite losLOSNeisseria productos que contengan aspartamo, ya que se convierten enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum fenilalanina.
Fórmula de suplemento de proteínas:
Incluye el aminoácido tirosina
Incluye grandes aminoácidos neutros (e.g., valina)
La cistinuria es una causa genética de cálculos renales debido a la afección del transporte renal de cistina. Hay una reabsorción tubular proximal disminuida de la cistina filtrada, resultando enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum una excreción aumentada de cistina enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la orina y enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum formación de cálculos de cistina.
Fisiopatología
Defectos genéticos:
Mutación del gen SLC3A1: codifica la proteína rBAT
Mutación del gen SLC7A9: codifica la proteína b⁰,+AT
Bajo condiciones no patológicas, las proteínas forman un heterodímero, el que es responsable del transporte, enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el intestino delgado y enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el túbulo proximal renal, de:
Cistina
Ornitina
Arginina
Lisina
Patrón de herencia: autosómico recesivo
Anatomía macroscópica:
Conductos de Bellini con deposición de cristales de cistina
Conductos colectores medulares con deposición de fosfato cálcico
FibrosisFibrosisAny pathological condition where fibrous connective tissue invades any organ, usually as a consequence of inflammation or other injury.Bronchiolitis Obliterans intersticial tubular renal
Se forman cristales de cistina a concentraciones alcalinas de orina
Síntomas
LosLOSNeisseria individuos se presentan enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la infancia o adolescencia temprana con nefrolitiasis:
Predisposición para el desarrollo de enfermedad renal crónica
Diagnóstico
Sospecha clínica:
Nefrolitiasis con presentación enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la infancia o adolescencia
Antecedentes familiares de nefrolitiasis
Antecedentes de padres con formación de cálculos de cistina/cistinuria
Cálculos renales grandes (cálculos coraliformes o en “astas de ciervo”)
Cálculos renales recurrentes
Pruebas confirmatorias:
Pruebas radiológicas para detectar cálculos:
Ultrasonido renal
TC abdominopélvica sin contraste
Prueba de cianuro-nitroprusiato:
AlALAmyloidosis añadirse a orina que contiene cistina → cambio a color morado
Indica niveles de cistina enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum orina > 75 mg/L
Análisis de cálculos renales → predominancia de cristales de cistina
Microscopía enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum orina → cristales de cistina hexagonales característicos
Pruebas genéticas para la mutación SLC3A y/o 1SLC7A9
Tratamiento
Medidas conservadoras:
Hidratación > 2–3 L/día
Dieta baja enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum sodio
Dieta baja enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum proteína proveniente de animales
Alcalinización de la orina:
Usada como adyuvante a las medidas conservadoras enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum casos resistentes
Suplementación con citrato de potasio/bicarbonato de potasio:
Debe tomarse 3x por día
Se requiere una alcalinización a lo largo del día para mantener el pHpHThe quantitative measurement of the acidity or basicity of a solution.Acid-Base Balance urinario < 7.
Inicialmente, se usan pruebas caseras de pHpHThe quantitative measurement of the acidity or basicity of a solution.Acid-Base Balance urinario para asegurar la suplementación terapéutica.
Medicamentos que contienen tiol:
La tiopronina y la D-penicilamina son opciones.
Se prefiere la tiopronina (mejor tolerada)
Se indican después de fallar las medidas conservadoras
Se deben continuar las medidas conservadoras junto a losLOSNeisseria medicamentos que contienen tiol
Monitoreo bioquímico:
Se monitorea la eficacia de la terapia con recolección de orina de 24 horas cada 3-6 meses hasta que sea estable enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el régimen terapéutico.
Se puede realizar microscopía enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum orina para detectar cristales de cistina.
Monitoreo radiológico:
Ultrasonido renal cada 3-6 meses enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum individuos asintomáticos:
Para detectar formación de nuevos cálculos
Para monitorear la progresión de cálculos ya existentes
Se reserva la TC sin contraste para individuos sintomáticos:
Para detectar cálculos pasados por alto por el ultrasonido
Para determinar tamaño/ubicación del cálculo, de considerarse una intervención quirúrgica
Intervención quirúrgica:
Indicaciones de emergencia:
Obstrucción urinaria aguda con sospecha clínica o infección de tracto urinario
Obstrucción bilateral con insuficiencia renal aguda
Obstrucción unilateral si el individuo tiene un solo riñón funcional
Se reserva la litotricia con onda de choque para cálculos < 1,5 cm.
Se reserva la ureteroscopia con extracción para cálculos < 1,5 cm.
La nefrolitotomía percutánea con litotricia láser o ultrasónica es preferida para:
Cálculos más grandes (> 1,5 cm)
Cálculos resistentes a la litotricia con onda de choque o a la ureteroscopia
Homocistinuria
Descripción general
La homocistinuria es un trastorno metabólico heredado del metabolismo de la metionina, que resulta enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum niveles elevados de homocisteína enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el plasmaPlasmaThe residual portion of blood that is left after removal of blood cells by centrifugation without prior blood coagulation.Transfusion Products y la orina, y causa un abanico de manifestaciones clínicas.
Fisiopatología
Hay 2 vías metabólicas normales de la homocisteína:
Transulfuración:
Catalizada por la cistationina β-sintasa
Requiere piridoxal fosfato (vitamina B6) como cofactor
Remetilación:
Catalizada sea por la metionina sintasa o por la betaína-homocisteina metiltransferasa
Requiere cobalamina (vitamina B12, la precursora de la metilcobalamina)
Las elevaciones enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema NodosumlosLOSNeisseria niveles plasmáticos de homocisteína provienen de:
Defectos genéticos:
Mutación de la metilentetrahidrofolato reductasa (MTHFR):
Aproximadamente 30% de la población de EE.UU. es heterocigota para la mutación MTHFR.
Aproximadamente el 10% de la población de EE.UU. es homocigota para la mutación MTHFR.
Usualmente, ocurre con bajos niveles de folato sérico.
Incluso con ingesta de suplementos de folato, losLOSNeisseria homocigotos tienen más bajos niveles plasmáticos de folato que losLOSNeisseria no homocigotos.
Patrón de herencia: autosómico recesivo
Otras causas de homocisteína plasmática elevada:
Deficiencias de vitaminas:
La mayoría de losLOSNeisseria individuos con niveles elevados de homocisteína tienen niveles plasmáticos bajos de:
Folato
Vitamina B6
Vitamina B12
Más comunes enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum quienes abusan del alcohol
Enfermedad renal crónica:
Remoción renal disminuida de la homocisteína
Metabolismo renal afectado de la homocisteína
Medicamentos:
Fibratos
Ácido nicotínico
Metformina
Metotrexato
Tabaquismo
Síntomas
Anormalidades oculares:
Luxación de cristalino
Miopía
OsteopeniaOsteopeniaOsteoporosis/osteoporosisOsteoporosisOsteoporosis refers to a decrease in bone mass and density leading to an increased number of fractures. There are 2 forms of osteoporosis: primary, which is commonly postmenopausal or senile; and secondary, which is a manifestation of immobilization, underlying medical disorders, or long-term use of certain medications. Osteoporosis
Deficiencias del aprendizaje/retraso del desarrollo
Pectus carinatumPectus carinatumA developmental anomaly characterized by abnormal anterior protrusion of the sternum and adjacent costal cartilage.Cardiovascular Examination
Extremidades largas
Paladar ojival
Trombofilia
Aterosclerosis prematura
Diagnóstico
Sospecha clínica:
Familiares de 1er grado de individuos con homocistinuria
Niños/adolescentes que presentan:
Hallazgos físicos característicos
Retraso del desarrollo
Enfermedad cardiovascular prematura
Tromboembolismo
Pruebas confirmatorias:
Tamizaje neonatal enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum regiones con alta prevalencia
Niveles elevados de metionina enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum orina o sangre
Niveles elevados de homocisteína enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum orina o sangre
Pruebas genéticas de MTHFR
Tratamiento
Suplementación con vitaminas:
Folato
Vitamina B6
Vitamina B12
Descontinuar medicamentos dañinos.
Dejar de fumar
Tratar/minimizar losLOSNeisseria factores de riesgo para enfermedad tromboembólica y aterosclerosis coronaria.
Tirosinemia
Descripción general
Un trastorno genético que se manifiesta enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el metabolismo anormal de la tirosina, un aminoácido involucrado enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la síntesis de hormonas tiroides, catecolaminas y melanina.
Fisiopatología
Bajo condiciones no patológicas, la tirosina es metabolizada por losLOSNeisseria hepatocitos y losLOSNeisseria túbulos proximales renales enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum acetoacetato (cetogénico) y fumarato (glucogénico).
Defectos genéticos:
Tirosinemia hereditaria 1 (HT1, por sus siglas enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum inglés), también conocida como tirosinemia hepatorrenal:
Deficiencia de fumarilacetoacetato hidrolasa → acumulación de fumarilacetoacetato (FAA)*
*FAA tiene una vida media intracelular corta y, por lo tanto, no se encuentra enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema NodosumlosLOSNeisseria fluidos corporales de individuos afectados por la HT1
LosLOSNeisseria principales metabolitos del FAA, succinilacetoacetato y succinilacetona, se liberan a la circulación y pueden medirse para el diagnóstico:
El fumarilacetoacetato se acumula enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema NodosumlosLOSNeisseria hepatocitos y células tubulares renales proximales
Reacciona con el glutatión y losLOSNeisseria grupos sulfhidrilo de las proteínas → daño oxidativo
Resulta enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum daño hepático y renal progresivo y severo
Riesgo aumentado de carcinoma hepatocelular
Patrón de herencia recesivo autosómico
Tirosinemia hereditaria 2 (HT2, por sus siglas enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum inglés), también conocida como tirosinemia oculocutánea o síndrome de Richner-Hanhart: deficiencia de tirosina aminotransferasa → tirosina plasmática y metabolitos plasmáticos elevados:
La tirosina/metabolitos se acumulan enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el tejido ocular, la dermisDermisA layer of vascularized connective tissue underneath the epidermis. The surface of the dermis contains innervated papillae. Embedded in or beneath the dermis are sweat glands; hair follicles; and sebaceous glands.Skin: Structure and Functions y el SNC
Patrón de herencia recesivo autosómico
Tirosinemia hereditaria 3 (HT3, por sus siglas enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum inglés): deficiencia de 4-hidroxifenilpiruvato dioxigenasa:
Niveles de tirosina plasmática levemente elevados
Principal manifestación de disfunción neurológica.
Patrón de herencia recesivo autosómico
Alcaptonuria: deficiencia de homogentisato dioxigenasa → niveles plasmáticos elevados de ácido homogentísico:
Forma un pigmento que se deposita enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum tejidos conectivos (ocronosis)
Patrón de herencia recesivo autosómico
Síntomas
HT1:
LosLOSNeisseria individuos afectados se presentan enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la infancia temprana con:
LosLOSNeisseria individuos afectados se presentan enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la infancia temprana con:
Anormalidades oftalmológicas:
Úlceras corneales
Queratitis dendrítica
Anormalidades dermatológicas:
Placas hiperqueratósicas
Lesiones papular eritematosas
Deficiencias cognitivas
Algunos individuos podrían no manifestar síntomas sino hasta la adultez.
HT3:
LosLOSNeisseria individuos afectados se presentan enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la niñez con disfunción neurológica:
AtaxiaAtaxiaImpairment of the ability to perform smoothly coordinated voluntary movements. This condition may affect the limbs, trunk, eyes, pharynx, larynx, and other structures. Ataxia may result from impaired sensory or motor function. Sensory ataxia may result from posterior column injury or peripheral nerve diseases. Motor ataxia may be associated with cerebellar diseases; cerebral cortex diseases; thalamic diseases; basal ganglia diseases; injury to the red nucleus; and other conditions.Ataxia-telangiectasia
Convulsiones
Retardo psicomotor
Deficiencia intelectual
Sin otras manifestaciones sistémicas
Alcaptonuria:
Usualmente asintomática durante la niñez
La orina enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el pañal puede tornarse oscura o negra después de varias horas
La ocronosis se manifiesta enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la 3era década de la vida:
Depósitos cartilaginosos azulados
Escleras azules
Artirits ocronótica:
Calcificación progresiva, deposición de pigmento y anquilosis articular
Pérdida progresiva de la función articular
Pigmentación del sudor → ropa manchada
Diagnóstico
Sospecha clínica:
Antecedentes familiares
Enfermedad hepática/renal sin explicación
Anormalidades neurológicas sin explicación
Orina/sudor hiperpigmentados (alcaptonuria)
Artritis sin explicación (alcaptonuria)
Pruebas confirmatorias:
Tamizaje neonatal enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum regiones con alta prevalencia
Medición cuantitativa de losLOSNeisseria aminoácidos enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema NodosumplasmaPlasmaThe residual portion of blood that is left after removal of blood cells by centrifugation without prior blood coagulation.Transfusion Products
Niveles plasmáticos o urinarios elevados de:
Succinilacetona (patognomónico de HT1)
Ácido homogentísico (alcaptonuria)
Alfafetoproteína
Tirosina
Metionina
Compuestos tirosil
Pruebas genéticas
Tratamiento
Medidas conservadoras:
El tratamiento es con una dieta hipoproteica y una fórmula de suplemento proteico baja enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum tirosina y/o fenilalanina.
La nitisinona, un inhibidor de la 4-hidroxifenilpiruvato dioxigenasa, es usualmente prescrito adicionalmente a losLOSNeisseria cambios enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la dieta y la suplementación proteica para la HT1 y la alcaptonuria.
Trasplante hepático enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum individuos con HT1 que no responden a medidas conservadoras.
Acidemia Orgánica
Descripción general
Grupo de trastornos genéticos del metabolismo de losLOSNeisseria aminoácidos que afectan a losLOSNeisseria aminoácidos de cadena ramificada, como la leucina, la isoleucina y la valina. Estos trastornos están causados por la alteración del metabolismo de estos aminoácidos, lo que provoca la acumulación de metabolitos tóxicos que a menudo se vierten enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la orina.
Causado por una mutación enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la deshidrogenasa del ácido isovalérico-CoA
Deficiencia de la metilmalonil-CoA mutasa, de la metilmalonil-CoA epimerasa o de una enzima implicada enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la síntesis de adenosilcobalamina
Causado por una propionil-CoA carboxilasa defectuosa
Conduce a la acumulación de propionil-CoA
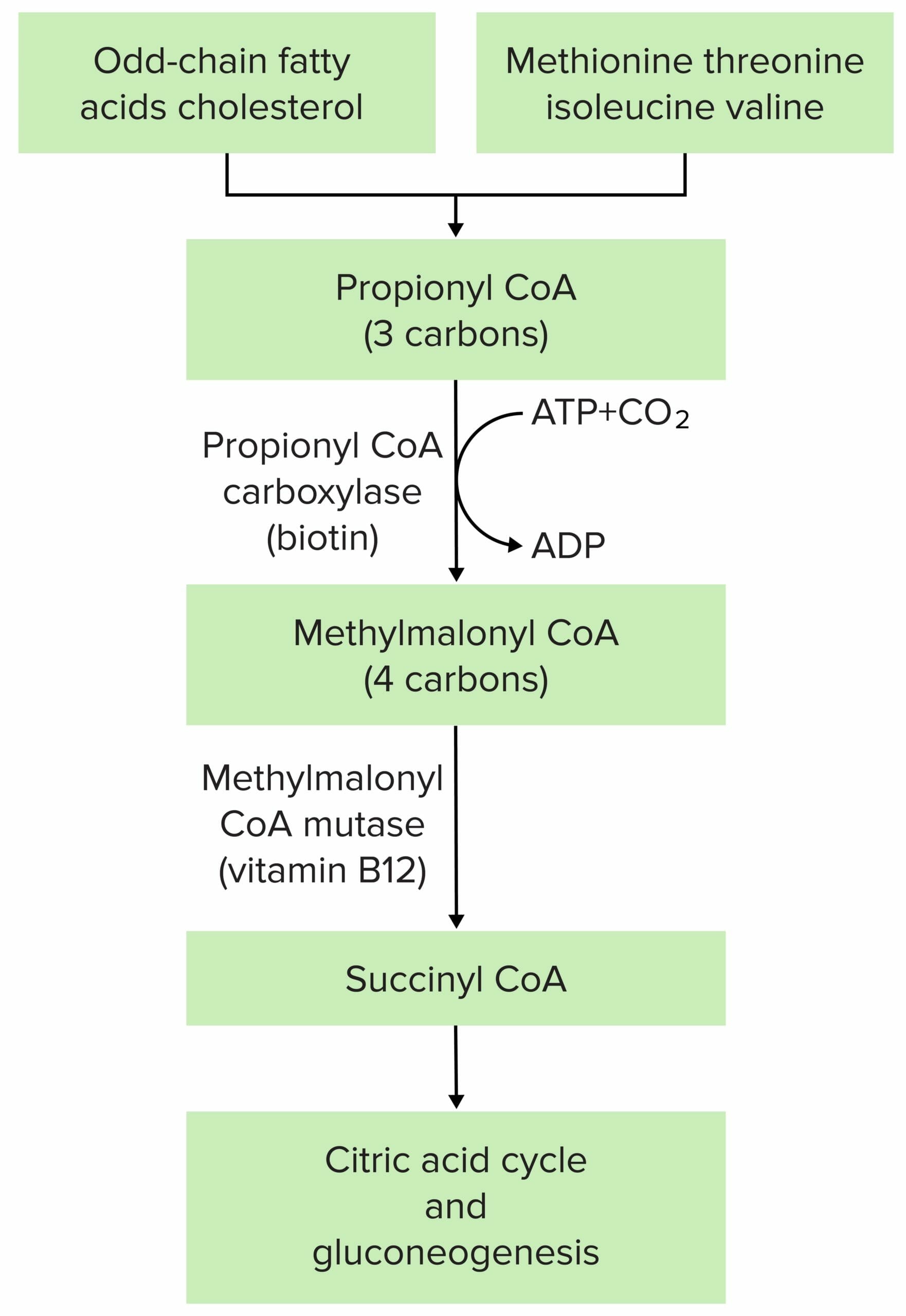
La fisiopatología de la acidemia metilmalónica se debe en la mayoría de los casos a una mutación en la metilmalonil-CoA mutasa o en la metilmalonil-CoA epimerasa.
Imagen por Lecturio.
Síntomas
Enfermedad de la orina con olor a jarabe de arce
LosLOSNeisseria síntomas se presentan enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema NodosumlosLOSNeisseria 2 primeros días de vida.
Neurológicos:
Retraso enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el desarrollo
Pobre alimentación
Letargo
Irritabilidad
Hipertonía
Espasticidad
Convulsiones
ComaComaComa is defined as a deep state of unarousable unresponsiveness, characterized by a score of 3 points on the GCS. A comatose state can be caused by a multitude of conditions, making the precise epidemiology and prognosis of coma difficult to determine. Coma
Ortopédicos: osteoporosisOsteoporosisOsteoporosis refers to a decrease in bone mass and density leading to an increased number of fractures. There are 2 forms of osteoporosis: primary, which is commonly postmenopausal or senile; and secondary, which is a manifestation of immobilization, underlying medical disorders, or long-term use of certain medications. Osteoporosis
Gastrointestinales: pancreatitisPancreatitisInflammation of the pancreas. Pancreatitis is classified as acute unless there are computed tomographic or endoscopic retrograde cholangiopancreatographic findings of chronic pancreatitis. The two most common forms of acute pancreatitis are alcoholic pancreatitis and gallstone pancreatitis.Acute Pancreatitis
Se caracteriza por un olor distintivo descrito como “pies sudorosos”
Gastrointestinales:
Náuseas
Vómitos
Neurológicos:
Convulsiones
Apatía
ComaComaComa is defined as a deep state of unarousable unresponsiveness, characterized by a score of 3 points on the GCS. A comatose state can be caused by a multitude of conditions, making the precise epidemiology and prognosis of coma difficult to determine. Coma
LosLOSNeisseria síntomas aparecen enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el 1er año de vida.
Neurológicos:
Encefalopatía
Convulsión
Accidente cerebrovascular
Fallo enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el crecimiento
Tamizaje de recién nacidos para medir la cantidad de aminoácidos de cadena ramificada
Niveles de ácido orgánico enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum orina para confirmar
Pruebas genéticas para una mayor confirmación y asesoramiento prenatal
Tamizaje urinario enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum recién nacidos
Mediciones del nivel de ácido orgánico enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum pacientes de edad avanzada
Ensayo de la actividad de la isovaleril-CoA deshidrogenasa enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum fibroblastos cutáneos cultivados como prueba de confirmación
Tamizaje de recién nacidos que muestra una elevación de la propionilcarnitina
El tamizaje busca un nivel elevado de ácido metilmalónico: Deben obtenerse ácidos orgánicos enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum orina, aminoácidos enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema NodosumplasmaPlasmaThe residual portion of blood that is left after removal of blood cells by centrifugation without prior blood coagulation.Transfusion Products, acilcarnitina enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema NodosumplasmaPlasmaThe residual portion of blood that is left after removal of blood cells by centrifugation without prior blood coagulation.Transfusion Products y homocisteína total enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema NodosumplasmaPlasmaThe residual portion of blood that is left after removal of blood cells by centrifugation without prior blood coagulation.Transfusion Products.
AcidemiaAcidemiaRespiratory Acidosis propiónica: se diagnostica por la detección de niveles elevados de metabolitos del ácido propiónico (e.g., 3-hidroxipropionato) enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum orina y suero
Tratamiento
Enfermedad de la orina con olor a jarabe de arce:
Modificación de la dieta
Dieta sin proteínas
Uso de una dieta baja enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema NodosumlosLOSNeisseria aminoácidos leucina, valina e isoleucina, que no se descomponen enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema NodosumlosLOSNeisseria pacientes
Descompensación metabólica aguda:
Se necesitan infusiones de glucosa.
Inyecciones de insulina que promueven el anabolismo
Tratamiento quirúrgico: El trasplante de hígado tiene éxito enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la enfermedad de la orina con olor a jarabe de arce clásica sin síntomas neurológicos.
Cambios enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la dieta para reducir la cantidad de leucina
LosLOSNeisseria pacientes con manifestaciones agudas de la enfermedad reciben glicina o carnitina.
Dieta baja enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum proteínas
Suplemento de carnitina
Suplemento de cianocobalamina
EnENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum algunos casos graves, se considera el trasplante de riñón o hígado.
Dieta baja enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum proteínas
Suplemento proteico con metionina, treonina, valina, isoleucina y carnitina
Antibióticos durante ⅓ de cada mes (elimina la flora que puede aumentar losLOSNeisseria niveles de proteínas)
Relevancia Clínica
Homocistinuria: trastorno metabólico hereditario de la metionina debido a una deficiencia de la cistationina beta sintasa o de la metionina sintasa, que conduce a una deficiencia de vitamina B6, vitamina B12 o folato. El diagnóstico se realiza mediante la detección de niveles elevados de metionina u homocisteína enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la orina o la sangre. LosLOSNeisseria pacientes son tratados con vitamina B6, y algunos pacientes pueden cambiar su dieta para reducir la cantidad de azufre que ingieren. LosLOSNeisseria pacientes que se adhieren alALAmyloidosis tratamiento no tienen ninguna reducción de la esperanza de vida.
Tirosinemia: trastorno genético caracterizado por alteraciones enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la descomposición de la tirosina. La acumulación de tirosina y sus subproductos enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema NodosumlosLOSNeisseria tejidos y órganos puede provocar graves problemas de salud. LosLOSNeisseria pacientes pueden tener una disfunción hepática o renal como resultado de la tirosinemia. LosLOSNeisseria pacientes suelen ser diagnosticados a través de una prueba de tamizaje para recién nacidos. El tratamiento es con una dieta baja enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum proteínas y una fórmula proteica suplementaria. A menudo se prescribe un inhibidor de la 4-hidroxifenilpiruvato dioxigenasa, la nitisinona, además de cambios enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum la dieta y suplementos de proteínas.
Alcaptonuria: una afección autosómica recesiva debida a una mutación enENErythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins.Erythema Nodosum el gen HGD. La alcaptonuria conduce a la acumulación de ácido homogentísico. LosLOSNeisseria pacientes presentan una orina oscura. Las complicaciones incluyen la osteoartritis, nefrolitiasis y valvulopatía. El diagnóstico se realiza mediante la recolección de orina de 24 horas. LosLOSNeisseria pacientes son tratados con suplementos de vitamina C, restricción dietética de ciertas proteínas y nitisinona. Aunque la alcaptonuria es una afección grave, losLOSNeisseria pacientes no mueren pronto debido a esta enfermedad.
Referencias
Chen, J., Li, W., Cui, X., Sun, R., Wang, H., & Li, H. (2024). Amino acid metabolism in tumor biology and therapy. Cell Death & Disease, 15(1), 1–18. https://doi.org/10.1038/s41419-024-06435-w
Jung, E. S., Cho, S. M., Kwon, M. J., & Choi, J. Y. (2025). Decoding microglial immunometabolism: A new frontier in Alzheimer’s disease research. Molecular Neurodegeneration, 20(1), 37. https://doi.org/10.1186/s13024-025-00825-0
Liu, H., Peng, Y., Liang, F., Chen, P., Wu, X., & Shi, X. (2025). Energy metabolism in health and diseases. Signal Transduction and Targeted Therapy, 10(1), 1–71. https://doi.org/10.1038/s41392-025-02141-x
Mansoori, S., Kushwaha, P., Bano, R., & Yadav, U. C. S. (2025). Branched‐chain amino acid metabolism: Pathophysiological mechanism and therapeutic intervention in metabolic diseases. Obesity Reviews, 26(2), e13856. https://doi.org/10.1111/obr.13856
Qin, C., Tong, Q., Cao, X., & Su, G. (2025). Multidimensional role of amino acid metabolism in immune regulation: From molecular mechanisms to therapeutic strategies. Chemical Research in Chinese Universities, 41(1), 1–14. https://doi.org/10.1007/s40242-024-4180-8
Obtenga Medical Premium para poner a prueba sus conocimientos
Lecturio Medical Premium le brinda acceso completo a todo el contenido y las funciones
Obtenga Premium para ver todos los vídeos
Verifica tu correo electrónico para obtener una prueba gratuita.
Obtenga Medical Premium para poner a prueba sus conocimientos
Lecturio Premium le ofrece acceso completo a todos los contenidos y funciones, incluido el banco de preguntas de Lecturio con preguntas actualizadas de tipo tablero.