Benign Benign Fibroadenoma bone Bone Bone is a compact type of hardened connective tissue composed of bone cells, membranes, an extracellular mineralized matrix, and central bone marrow. The 2 primary types of bone are compact and spongy. Bones: Structure and Types tumors are a group of noncancerous neoplasms which form from bone Bone Bone is a compact type of hardened connective tissue composed of bone cells, membranes, an extracellular mineralized matrix, and central bone marrow. The 2 primary types of bone are compact and spongy. Bones: Structure and Types or cartilage Cartilage Cartilage is a type of connective tissue derived from embryonic mesenchyme that is responsible for structural support, resilience, and the smoothness of physical actions. Perichondrium (connective tissue membrane surrounding cartilage) compensates for the absence of vasculature in cartilage by providing nutrition and support. Cartilage: Histology. These lesions have distinct features, such as endosteal reactions with intraosseous calcification and bone Bone Bone is a compact type of hardened connective tissue composed of bone cells, membranes, an extracellular mineralized matrix, and central bone marrow. The 2 primary types of bone are compact and spongy. Bones: Structure and Types formation. There are several common benign Benign Fibroadenoma bone Bone Bone is a compact type of hardened connective tissue composed of bone cells, membranes, an extracellular mineralized matrix, and central bone marrow. The 2 primary types of bone are compact and spongy. Bones: Structure and Types tumors; osteoid Osteoid Bones: Development and Ossification osteoma, osteoblastoma, osteochondroma, osteoma, and giant-cell tumors are discussed here. Diagnosis is made on x-ray X-ray Penetrating electromagnetic radiation emitted when the inner orbital electrons of an atom are excited and release radiant energy. X-ray wavelengths range from 1 pm to 10 nm. Hard x-rays are the higher energy, shorter wavelength x-rays. Soft x-rays or grenz rays are less energetic and longer in wavelength. The short wavelength end of the x-ray spectrum overlaps the gamma rays wavelength range. The distinction between gamma rays and x-rays is based on their radiation source. Pulmonary Function Tests, which shows localized lesions in the bone Bone Bone is a compact type of hardened connective tissue composed of bone cells, membranes, an extracellular mineralized matrix, and central bone marrow. The 2 primary types of bone are compact and spongy. Bones: Structure and Types with sharp margins and without soft tissue Soft Tissue Soft Tissue Abscess involvement. A biopsy Biopsy Removal and pathologic examination of specimens from the living body. Ewing Sarcoma may also be necessary. Asymptomatic lesions require no management, but surgical removal or curettage may be required for symptomatic lesions and/or to prevent pathologic fractures.
Last updated: Dec 15, 2025
Osteoid Osteoid Bones: Development and Ossification osteomas Osteomas A benign tumor composed of bone tissue or a hard tumor of bonelike structure developing on a bone (homoplastic osteoma) or on other structures (heteroplastic osteoma). Familial Adenomatous Polyposis and osteoblastomas are bone-forming lesions found in children; osteomas Osteomas A benign tumor composed of bone tissue or a hard tumor of bonelike structure developing on a bone (homoplastic osteoma) or on other structures (heteroplastic osteoma). Familial Adenomatous Polyposis can also be seen in adults. Osteochondromas are cartilage-forming tumors. Giant-cell tumors are osteolytic. Fibromas and bone Bone Bone is a compact type of hardened connective tissue composed of bone cells, membranes, an extracellular mineralized matrix, and central bone marrow. The 2 primary types of bone are compact and spongy. Bones: Structure and Types cysts Cysts Any fluid-filled closed cavity or sac that is lined by an epithelium. Cysts can be of normal, abnormal, non-neoplastic, or neoplastic tissues. Fibrocystic Change are other benign Benign Fibroadenoma bone lesions Bone Lesions Multiple Myeloma.
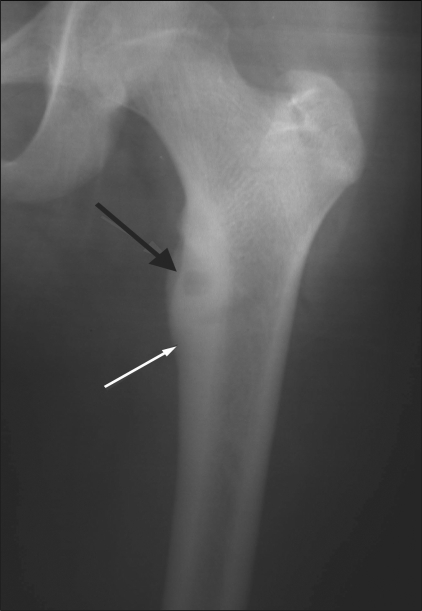
X-ray showing cortical osteoid osteoma involving the upper end of the femur:
The nidus is well seen (black arrow) along with the surrounding cortical thickening (white arrow).
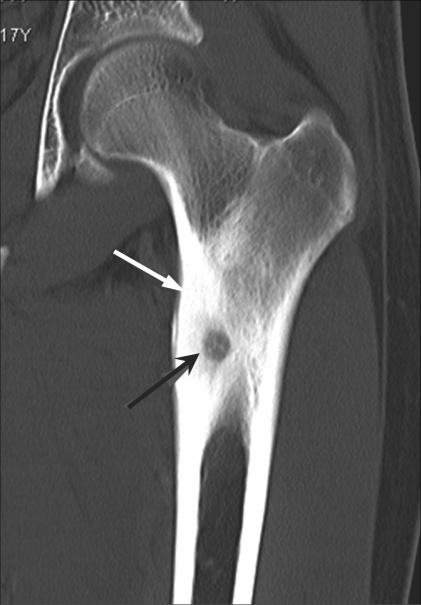
Coronal CT confirms the radiolucent nidus (black arrow) with surrounding bony sclerosis and cortical thickening (white arrow) in an osteoid osteoma.
Image: “Osteoid osteoma of the femur” by Jankharia B, Burute N. License: CC BY 2.0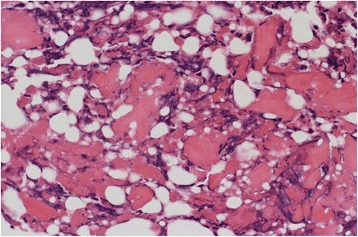
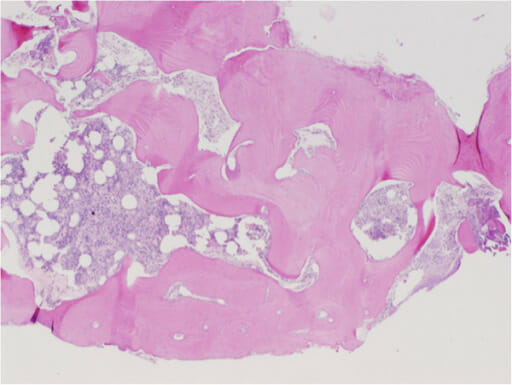
Histological findings show random reticular osteoid formation within a fibrovascular stroma, consistent with osteoid osteoma.
Image: “Histological findings. Histological findings showing random reticular osteoid formation within a fibrovascular stroma, consistent with osteoid osteoma” by Kazutaka Kikuta et al. License: CC BY 4.0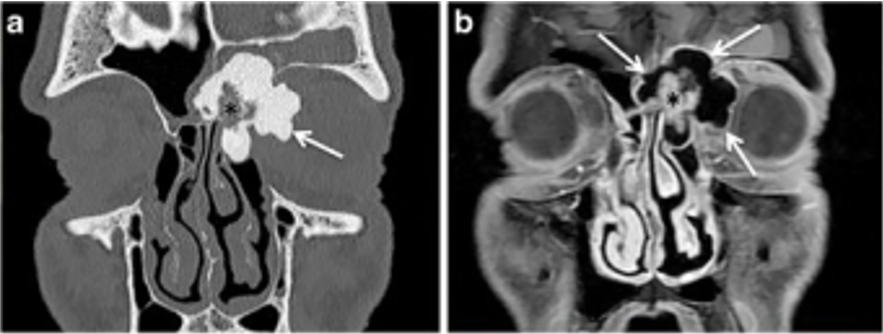
Histologically proven case of ethmoid sinus osteoma in a 15-year-old boy with left orbital pain and recurrent sinusitis for 6 months
a: Coronal CT scan (bone window) shows a well-circumscribed mass (arrow) with ossification of variable density. The peripheral dense portion corresponds to ivory osteoma (arrow), whereas the central portion (asterisk) corresponds to trabecular osteoma.
b: Corresponding coronal fat-saturated T1-weighted image obtained after IV gadolinium. The central osteoma portion (trabecular bone with marrow) enhances after contrast material (asterisk). In the absence of CT, the ivory osteoma portion can be misinterpreted as air.
Histopathological section of a middle ear osteoma shows fragments of lamellar bone with normal marrow elements.
Image: “Histopathological section of middle ear osteoma showing fragments of lamellar bone with normal marrow elements (stained with hematoxylin and eosin).” by Kate Curtis et al. License: CC BY 4.0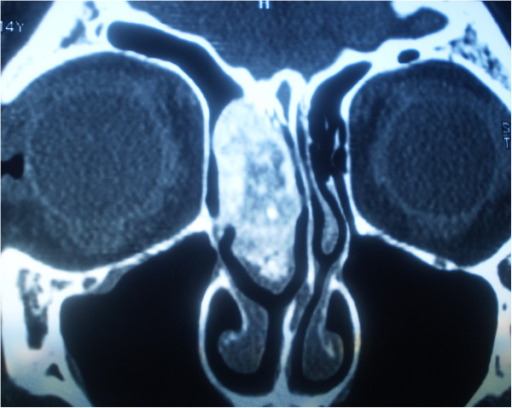
Coronal CT image of an osteoblastoma
Image: “Coronal multislice computed tomography image of osteoblastoma” by Cikojević D, Colović Z, Lozić B, Klančnik M. License: CC BY 2.0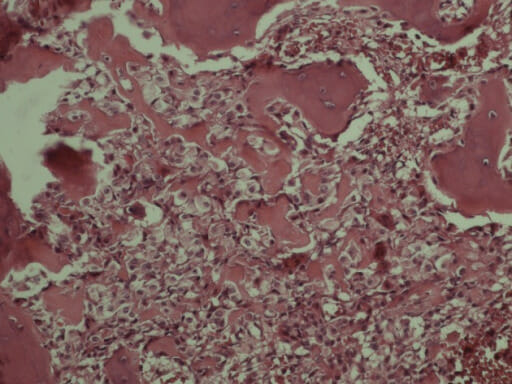
Variable osteoid calcifications lined by plump osteoblasts are visible and characteristic of osteoblastoma.
Image: “Variable osteoid calcifications lined by plump osteoblasts are also visible that are characteristic of osteoblastoma (×20).” by Shokouh Taghipour Zahi et al. License: CC BY 3.0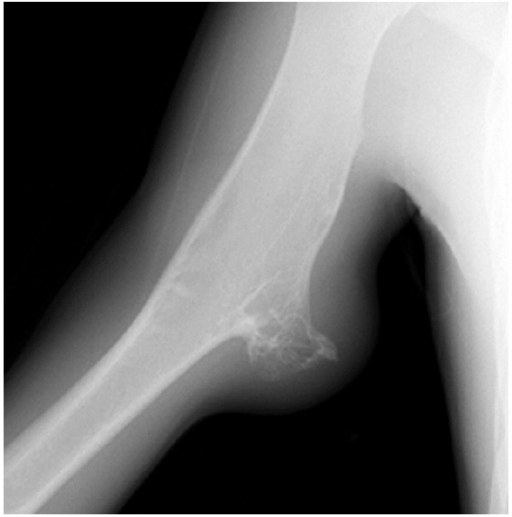
Oblique radiograph of the right arm shows soft tissue swelling overlying a broad-based osteochondroma arising from the humerus and directed toward the chest wall.
Image: “Oblique radiograph of the right arm” by Maras Ozdemır Z, Karakaplan M, Kahraman AS, Karadag N. License: CC BY 3.0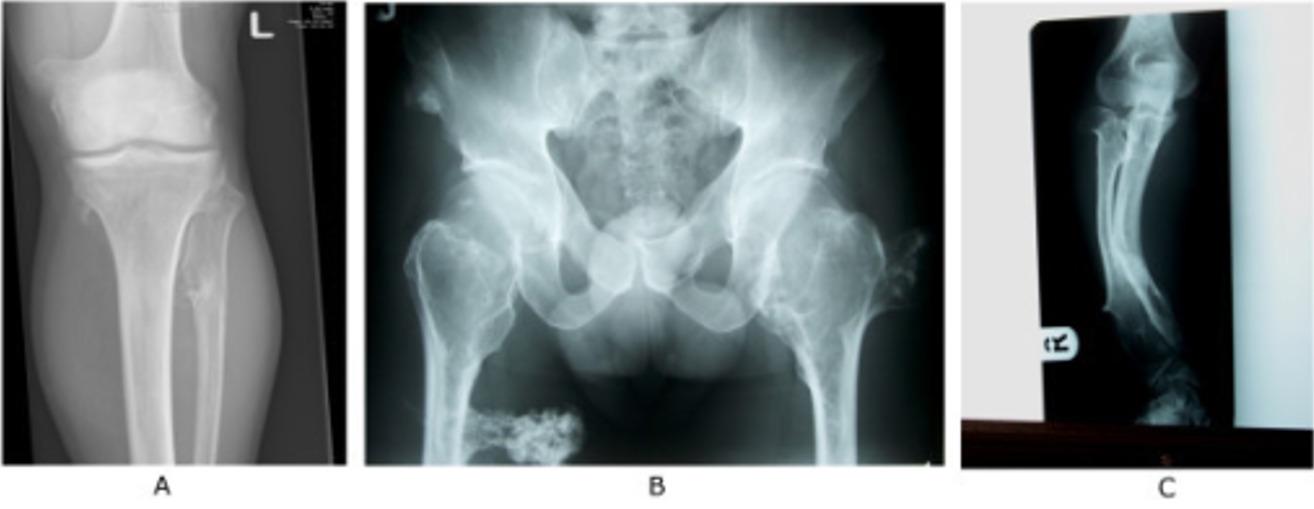
Examples of radiographs demonstrating multiple osteochondromas around the knee (A) and at the pelvis and proximal femur (B); panel C demonstrates the deformity of the forearm (shortening of the ulna with secondary bowing of radius) that is found in 39%–60% of individuals.
Image: “Examples of radiographs demonstrating multiple osteochondromas” by Bovée JV. License: CC BY 2.0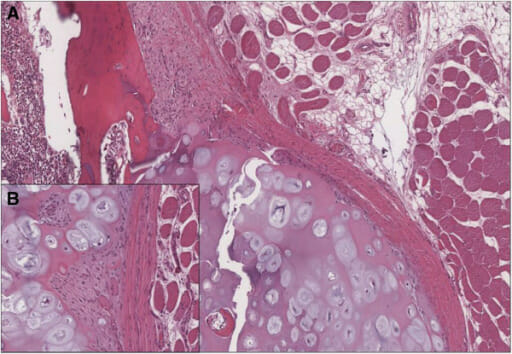
Histological examination confirmed the clinical diagnosis of osteochondroma:
A) Cartilage-capped trabecular bone covered by fibrous perichondrial tissue is visible.
B) The cartilaginous cap of osteochondroma shows no evidence of malignant transformation.
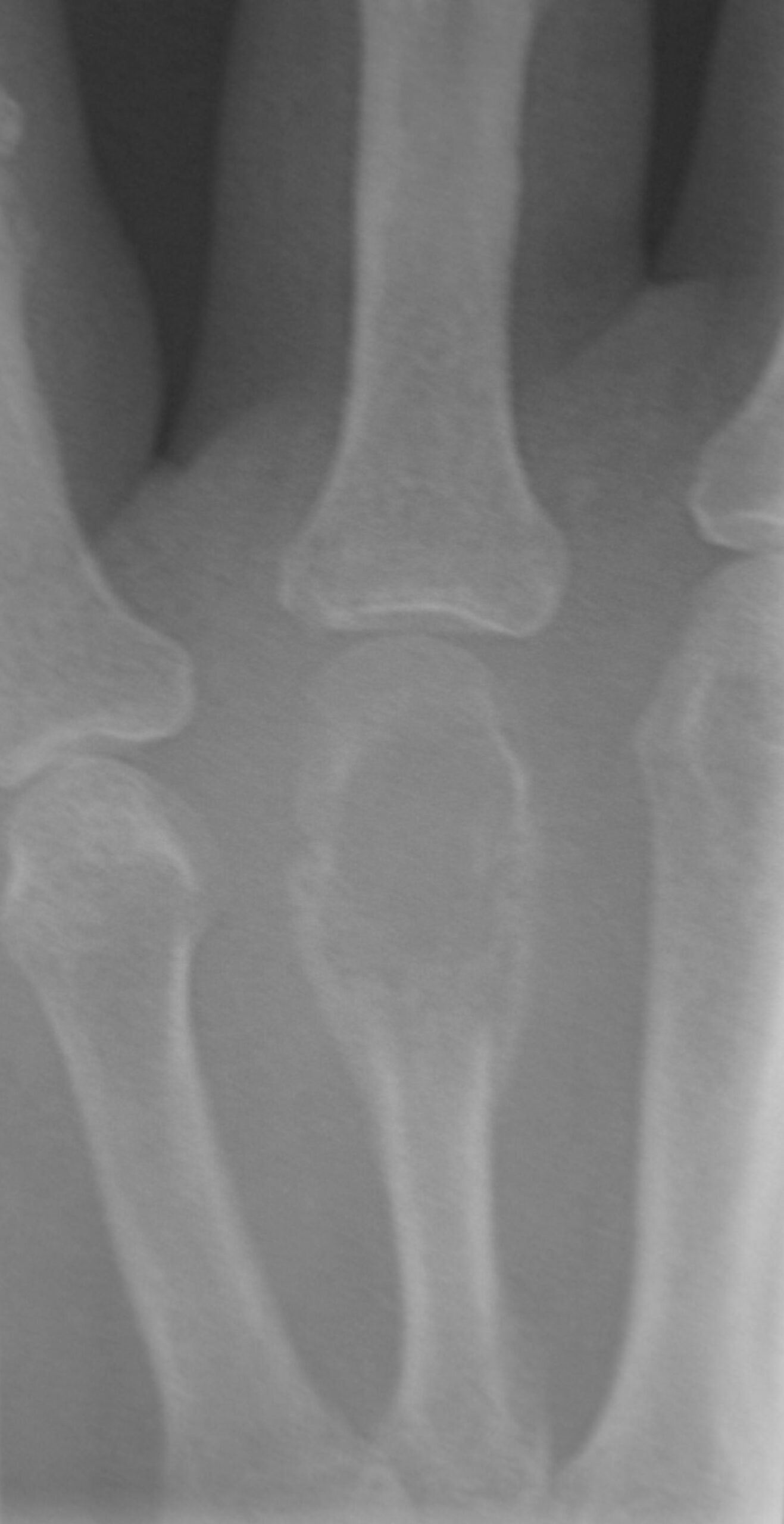
“Soap bubble” appearance of giant cell tumor in 4th metatarsal
Image: “Giant cell tumor of bone08” by James Heilman. License: Public Domain
Giant-cell tumor of the left distal femur:
A: X-ray film reveals osteolytic destruction and cortical thinning at the left femoral condyle.
B and C: T1-weighted MRI reveals low signal at the distal femur and a lateral visible tumor penetrating the front side of the cortex.
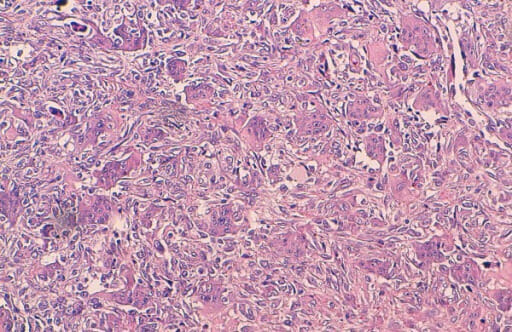
Histological findings of a giant cell tumor of the bone:
The tumor is composed of round, mononuclear stromal cells and uniformly scattered multinucleated giant cells (many contain a large number of nuclei). Characteristically, the nuclei of both stromal and giant cells are very similar.