Craniopharyngiomas are rare intracranial squamous epithelial tumors with a solid and/or cysticCysticFibrocystic Change structure that arise from the remnants of Rathke’s pouch along the pituitaryPituitaryA small, unpaired gland situated in the sella turcica. It is connected to the hypothalamus by a short stalk which is called the infundibulum.Hormones: Overview and Types stalk, in the suprasellar region. Craniopharyngiomas are histologically benignBenignFibroadenoma but tend to invade surrounding structures; thus, they should be treated as low-grade malignancies. Histologically, there are 2 types of tumors: adamantinomatous, which is more common in children; and papillary, which tends to occur in adults. Both these types can present with various symptoms depending on their location and extensionExtensionExamination of the Upper Limbs patterns. The symptoms include headaches, nauseaNauseaAn unpleasant sensation in the stomach usually accompanied by the urge to vomit. Common causes are early pregnancy, sea and motion sickness, emotional stress, intense pain, food poisoning, and various enteroviruses.Antiemetics, vomitingVomitingThe forcible expulsion of the contents of the stomach through the mouth.Hypokalemia, visual disturbances, endocrine dysfunction, and behavioral issues. Diagnosis is made by imaging and histology. Management typically involves surgical excision and radiationRadiationEmission or propagation of acoustic waves (sound), electromagnetic energy waves (such as light; radio waves; gamma rays; or x-rays), or a stream of subatomic particles (such as electrons; neutrons; protons; or alpha particles).Osteosarcoma therapy.
Craniopharyngiomas are rare squamous epithelial tumors with a solid and/or cysticCysticFibrocystic Change structure, which arise from the remnants of Rathke’s pouch along the pituitaryPituitaryA small, unpaired gland situated in the sella turcica. It is connected to the hypothalamus by a short stalk which is called the infundibulum.Hormones: Overview and Types stalk, in line from the nasopharynxNasopharynxThe top portion of the pharynx situated posterior to the nose and superior to the soft palate. The nasopharynx is the posterior extension of the nasal cavities and has a respiratory function.Pharynx: Anatomy to the diencephalonDiencephalonThe paired caudal parts of the prosencephalon from which the thalamus; hypothalamus; epithalamus; and subthalamus are derived.Development of the Nervous System and Face (in the suprasellar region).
Craniopharyngiomas have benignBenignFibroadenoma histology but malignant behavior, meaning that they tend to invade the surrounding structures and can shorten life expectancyLife expectancyBased on known statistical data, the number of years which any person of a given age may reasonably expected to live.Population Pyramids. Therefore, despite their benignBenignFibroadenoma appearance on histology, they are considered low-grade malignancies.
Classification of nervous systemNervous systemThe nervous system is a small and complex system that consists of an intricate network of neural cells (or neurons) and even more glial cells (for support and insulation). It is divided according to its anatomical components as well as its functional characteristics. The brain and spinal cord are referred to as the central nervous system, and the branches of nerves from these structures are referred to as the peripheral nervous system.Nervous System: Anatomy, Structure, and Classification tumors
Table: Classification of nervous systemNervous systemThe nervous system is a small and complex system that consists of an intricate network of neural cells (or neurons) and even more glial cells (for support and insulation). It is divided according to its anatomical components as well as its functional characteristics. The brain and spinal cord are referred to as the central nervous system, and the branches of nerves from these structures are referred to as the peripheral nervous system.Nervous System: Anatomy, Structure, and Classification tumors
Categories
Specific tumors
Neuroepithelial tumors in the CNS
Astrocytomas, including glioblastoma multiformeGlioblastoma multiformeGlioblastoma multiforme is a high-grade astrocytoma, an aggressive brain tumor arising from astrocytes, with an unknown cause and a poorly understood link to risk factors. There are two main types: primary, a more aggressive form seen more commonly in older patients, and secondary, developing from lower-grade astrocytomas and seen more commonly in younger patients.Glioblastoma Multiforme
OligodendrogliomaOligodendrogliomaOligodendrogliomas are malignant CNS tumors arising from neural glial cell precursors. Oligodendrogliomas often arise in the frontal lobes of the brain and have a generally favorable prognosis when compared to other gliomas. Oligodendrogliomas are the 3rd most common CNS tumor. The most frequent presenting symptom is a seizure.Oligodendroglioma
EpendymomaEpendymomaEpendymomas are glial cell tumors arising from CSF-producing ependymal cells lining the ventricular system. Ependymomas most commonly occur within the posterior fossa in contact with the 4th ventricle, or within the intramedullary spinal cord. Ependymoma and choroidChoroidThe thin, highly vascular membrane covering most of the posterior of the eye between the retina and sclera.Eye: Anatomy plexus tumors
Medulloblastomas (embryonal tumors)
Meningeal tumors
Meningiomas
HemangioblastomasHemangioblastomasA benign tumor of the nervous system that may occur sporadically or in association with von Hippel-Lindau disease. It accounts for approximately 2% of intracranial tumors, arising most frequently in the cerebellar hemispheres and vermis. Histologically, the tumors are composed of multiple capillary and sinusoidal channels lined with endothelial cells and clusters of lipid-laden pseudoxanthoma cells. Usually solitary, these tumors can be multiple and may also occur in the brain stem, spinal cord, retina, and supratentorial compartment. Cerebellar hemangioblastomas usually present in the third decade with intracranial hypertension, and ataxia.Von Hippel-Lindau Disease
Sellar region tumors
Craniopharyngioma
PituitaryPituitaryA small, unpaired gland situated in the sella turcica. It is connected to the hypothalamus by a short stalk which is called the infundibulum.Hormones: Overview and Types adenoma
MetastasisMetastasisThe transfer of a neoplasm from one organ or part of the body to another remote from the primary site.Grading, Staging, and Metastasis to the brainBrainThe part of central nervous system that is contained within the skull (cranium). Arising from the neural tube, the embryonic brain is comprised of three major parts including prosencephalon (the forebrain); mesencephalon (the midbrain); and rhombencephalon (the hindbrain). The developed brain consists of cerebrum; cerebellum; and other structures in the brain stem.Nervous System: Anatomy, Structure, and Classification (5x more common than primary brainBrainThe part of central nervous system that is contained within the skull (cranium). Arising from the neural tube, the embryonic brain is comprised of three major parts including prosencephalon (the forebrain); mesencephalon (the midbrain); and rhombencephalon (the hindbrain). The developed brain consists of cerebrum; cerebellum; and other structures in the brain stem.Nervous System: Anatomy, Structure, and Classification tumors)
Most commonly arising from:
Lung, breast, and renal cell carcinomas
MelanomaMelanomaMelanoma is a malignant tumor arising from melanocytes, the melanin-producing cells of the epidermis. These tumors are most common in fair-skinned individuals with a history of excessive sun exposure and sunburns. Melanoma
Peripheral tumors
Schwannomas, including acoustic neuromaAcoustic neuromaAcoustic neuroma, also referred to as vestibular schwannoma, is a benign tumor arising from Schwann cells of the vestibular component of the cranial nerve VIII. Acoustic neuroma forms within the internal auditory meatus and extends into the cerebellopontine angle. Acoustic Neuroma
NeuroblastomaNeuroblastomaNeuroblastoma is a malignancy that arises from the neural crest cell derivatives along the sympathetic chain (neuroblasts) and is most commonly located in the adrenal medulla. The tumor often presents in childhood with a flank mass that crosses the midline.Neuroblastoma
Epidemiology
IncidenceIncidenceThe number of new cases of a given disease during a given period in a specified population. It also is used for the rate at which new events occur in a defined population. It is differentiated from prevalence, which refers to all cases in the population at a given time.Measures of Disease Frequency: rare
Approximately 2 cases per 1,000,000 person years
Constitute 1%–3% of all primary brainBrainThe part of central nervous system that is contained within the skull (cranium). Arising from the neural tube, the embryonic brain is comprised of three major parts including prosencephalon (the forebrain); mesencephalon (the midbrain); and rhombencephalon (the hindbrain). The developed brain consists of cerebrum; cerebellum; and other structures in the brain stem.Nervous System: Anatomy, Structure, and Classification tumors
Race/ethnicity: more common in patientsPatientsIndividuals participating in the health care system for the purpose of receiving therapeutic, diagnostic, or preventive procedures.Clinician–Patient Relationship of Japanese and African descent
Age and subtype: A bimodal age distribution is observed.
Children:
Peak diagnosis at 5–14 years of age
More commonly adamantinomatous subtype
Adults:
Peak diagnosis at 50–75 years of age
More commonly the papillary subtype
High recurrence rate (50%)
High morbidityMorbidityThe proportion of patients with a particular disease during a given year per given unit of population.Measures of Health Status rate
Headaches
Visual symptoms
Hormonal disturbances
ObesityObesityObesity is a condition associated with excess body weight, specifically with the deposition of excessive adipose tissue. Obesity is considered a global epidemic. Major influences come from the western diet and sedentary lifestyles, but the exact mechanisms likely include a mixture of genetic and environmental factors. Obesity
Mental health disturbances
Classification
There are 2 primary types of craniopharyngiomas:
Adamantinomatous type:
Arise from the embryonic cells of the craniopharyngeal duct
Arise from the cells of the anterior portion of the pituitaryPituitaryA small, unpaired gland situated in the sella turcica. It is connected to the hypothalamus by a short stalk which is called the infundibulum.Hormones: Overview and Types gland
Well circumscribed, solid or cysticCysticFibrocystic Change, and filled with a viscous yellow fluid
Craniopharyngiomas are squamous epithelial tumors that arise along the pituitaryPituitaryA small, unpaired gland situated in the sella turcica. It is connected to the hypothalamus by a short stalk which is called the infundibulum.Hormones: Overview and Types stalk in the suprasellar region, adjacent to the optic chiasmOptic ChiasmThe x-shaped structure formed by the meeting of the two optic nerves. At the optic chiasm the fibers from the medial part of each retina cross to project to the other side of the brain while the lateral retinal fibers continue on the same side. As a result each half of the brain receives information about the contralateral visual field from both eyes.The Visual Pathway and Related Disorders.
Etiology
Two main hypotheses have been proposed to explain the etiology of craniopharyngiomas:
Embryonic theory:
Postulates that embryonic cells from the Rathke’s cleft and the remnants of the craniopharyngeal duct are the site of origin
The more likely etiology in the adamantinomatous type, which is more common in children
Metaplastic theory:
Postulates that residual, mature squamous cells of the anterior hypophysisHypophysisThe pituitary gland, also known as the hypophysis, is considered the “master endocrine gland” because it releases hormones that regulate the activity of multiple major endocrine organs in the body. The gland sits on the sella turcica, just below the hypothalamus, which is the primary regulator of the pituitary gland.Pituitary Gland: Anatomy undergo metaplasiaMetaplasiaA condition in which there is a change of one adult cell type to another similar adult cell type.Cellular Adaptation
The more likely etiology in the papillary type, which is more common in adults
Adamantinomatous type: CTNNB1 geneGeneA category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms.Basic Terms of Genetics mutations → mutations in β-catenin protein
Involved in patterning decisions during embryonic development, cell proliferation, and cell adhesionAdhesionThe process whereby platelets adhere to something other than platelets, e.g., collagen; basement membrane; microfibrils; or other ‘foreign’ surfaces.Coagulation Studies
Papillary type: activating mutations in the BRAF V600E oncogene → inhibition of apoptosisApoptosisA regulated cell death mechanism characterized by distinctive morphologic changes in the nucleus and cytoplasm, including the endonucleolytic cleavage of genomic DNA, at regularly spaced, internucleosomal sites, I.e., DNA fragmentation. It is genetically-programmed and serves as a balance to mitosis in regulating the size of animal tissues and in mediating pathologic processes associated with tumor growth.Ischemic Cell Damage/enhanced cell survival
Arise in the pituitaryPituitaryA small, unpaired gland situated in the sella turcica. It is connected to the hypothalamus by a short stalk which is called the infundibulum.Hormones: Overview and Types stalk in the suprasellar region
Project into the hypothalamusHypothalamusThe hypothalamus is a collection of various nuclei within the diencephalon in the center of the brain. The hypothalamus plays a vital role in endocrine regulation as the primary regulator of the pituitary gland, and it is the major point of integration between the central nervous and endocrine systems.Hypothalamus
May extend horizontally along the path of least resistanceResistancePhysiologically, the opposition to flow of air caused by the forces of friction. As a part of pulmonary function testing, it is the ratio of driving pressure to the rate of air flow.Ventilation: Mechanics of Breathing, which may be anteriorly, posteriorly, and/or laterally:
Clinical presentation will depend on the affected areas of the brainBrainThe part of central nervous system that is contained within the skull (cranium). Arising from the neural tube, the embryonic brain is comprised of three major parts including prosencephalon (the forebrain); mesencephalon (the midbrain); and rhombencephalon (the hindbrain). The developed brain consists of cerebrum; cerebellum; and other structures in the brain stem.Nervous System: Anatomy, Structure, and Classification. Commonly affected areas include:
Anterior pituitaryPituitaryA small, unpaired gland situated in the sella turcica. It is connected to the hypothalamus by a short stalk which is called the infundibulum.Hormones: Overview and Types (and its hormonesHormonesHormones are messenger molecules that are synthesized in one part of the body and move through the bloodstream to exert specific regulatory effects on another part of the body. Hormones play critical roles in coordinating cellular activities throughout the body in response to the constant changes in both the internal and external environments. Hormones: Overview and Types)
Optic chiasmOptic ChiasmThe x-shaped structure formed by the meeting of the two optic nerves. At the optic chiasm the fibers from the medial part of each retina cross to project to the other side of the brain while the lateral retinal fibers continue on the same side. As a result each half of the brain receives information about the contralateral visual field from both eyes.The Visual Pathway and Related Disorders and/or optic nerves
Craniopharyngiomas are slow-growing tumors; therefore, there is an insidious development of symptoms usually after the tumorTumorInflammation attains a diameter > 3 cm. The onset of symptoms is usually 1‒2 years after the tumorTumorInflammation develops.
General symptoms
HeadacheHeadacheThe symptom of pain in the cranial region. It may be an isolated benign occurrence or manifestation of a wide variety of headache disorders.Brain Abscess:
Most common presentation, seen in 50%‒80% of cases
Presentation is progressive, dull, continuous, and positional.
LethargyLethargyA general state of sluggishness, listless, or uninterested, with being tired, and having difficulty concentrating and doing simple tasks. It may be related to depression or drug addiction.Hyponatremia
NauseaNauseaAn unpleasant sensation in the stomach usually accompanied by the urge to vomit. Common causes are early pregnancy, sea and motion sickness, emotional stress, intense pain, food poisoning, and various enteroviruses.Antiemetics and/or vomitingVomitingThe forcible expulsion of the contents of the stomach through the mouth.Hypokalemia
Visual disturbances
Visual disturbances are seen in about 40%‒65% of patientsPatientsIndividuals participating in the health care system for the purpose of receiving therapeutic, diagnostic, or preventive procedures.Clinician–Patient Relationship and include:
DiplopiaDiplopiaA visual symptom in which a single object is perceived by the visual cortex as two objects rather than one. Disorders associated with this condition include refractive errors; strabismus; oculomotor nerve diseases; trochlear nerve diseases; abducens nerve diseases; and diseases of the brain stem and occipital lobe.Myasthenia Gravis
Decrease in visual acuityVisual AcuityClarity or sharpness of ocular vision or the ability of the eye to see fine details. Visual acuity depends on the functions of retina, neuronal transmission, and the interpretative ability of the brain. Normal visual acuity is expressed as 20/20 indicating that one can see at 20 feet what should normally be seen at that distance. Visual acuity can also be influenced by brightness, color, and contrast.Ophthalmic Exam
Some type of endocrine dysfunction is common in craniopharyngiomas, and is seen in about 65%‒90% of patientsPatientsIndividuals participating in the health care system for the purpose of receiving therapeutic, diagnostic, or preventive procedures.Clinician–Patient Relationship. Deficiencies in growth hormone (GH), gonadotropins, thyroid-stimulating hormoneThyroid-stimulating hormoneA glycoprotein hormone secreted by the adenohypophysis. Thyrotropin stimulates thyroid gland by increasing the iodide transport, synthesis and release of thyroid hormones (thyroxine and triiodothyronine).Thyroid Hormones (TSH), and adrenocorticotropic hormoneAdrenocorticotropic hormoneAn anterior pituitary hormone that stimulates the adrenal cortex and its production of corticosteroids. Acth is a 39-amino acid polypeptide of which the n-terminal 24-amino acid segment is identical in all species and contains the adrenocorticotropic activity. Upon further tissue-specific processing, acth can yield alpha-msh and corticotropin-like intermediate lobe peptide (clip).Adrenal Hormones (ACTH) can all be seen.
GH deficiencies:
In young patientsPatientsIndividuals participating in the health care system for the purpose of receiving therapeutic, diagnostic, or preventive procedures.Clinician–Patient Relationship: growth failureGrowth failureChronic Granulomatous Disease and delayed pubertyDelayed PubertyDelayed puberty (DP) is defined as the lack of testicular growth in boys past the age of 14 and the lack of thelarche in girls past the age of 13. Delayed puberty affects up to 5% of healthy boys and girls, and half of all cases are due to constitutional growth delay.Delayed Puberty
In adults: obesityObesityObesity is a condition associated with excess body weight, specifically with the deposition of excessive adipose tissue. Obesity is considered a global epidemic. Major influences come from the western diet and sedentary lifestyles, but the exact mechanisms likely include a mixture of genetic and environmental factors. Obesity, fatigueFatigueThe state of weariness following a period of exertion, mental or physical, characterized by a decreased capacity for work and reduced efficiency to respond to stimuli.Fibromyalgia
HypothyroidismHypothyroidismHypothyroidism is a condition characterized by a deficiency of thyroid hormones. Iodine deficiency is the most common cause worldwide, but Hashimoto’s disease (autoimmune thyroiditis) is the leading cause in non-iodine-deficient regions. Hypothyroidism(↓ TSH, approximately 40% of patientsPatientsIndividuals participating in the health care system for the purpose of receiving therapeutic, diagnostic, or preventive procedures.Clinician–Patient Relationship):
Weight gain/obesityObesityObesity is a condition associated with excess body weight, specifically with the deposition of excessive adipose tissue. Obesity is considered a global epidemic. Major influences come from the western diet and sedentary lifestyles, but the exact mechanisms likely include a mixture of genetic and environmental factors. Obesity
Cold intolerance
ConstipationConstipationConstipation is common and may be due to a variety of causes. Constipation is generally defined as bowel movement frequency < 3 times per week. Patients who are constipated often strain to pass hard stools. The condition is classified as primary (also known as idiopathic or functional constipation) or secondary, and as acute or chronic. Constipation
FatigueFatigueThe state of weariness following a period of exertion, mental or physical, characterized by a decreased capacity for work and reduced efficiency to respond to stimuli.Fibromyalgia
Adrenal dysfunction (↓ ACTH, approximately 25% of patientsPatientsIndividuals participating in the health care system for the purpose of receiving therapeutic, diagnostic, or preventive procedures.Clinician–Patient Relationship):
Orthostatic hypotensionOrthostatic hypotensionA significant drop in blood pressure after assuming a standing position. Orthostatic hypotension is a finding, and defined as a 20-mm hg decrease in systolic pressure or a 10-mm hg decrease in diastolic pressure 3 minutes after the person has risen from supine to standing. Symptoms generally include dizziness, blurred vision, and syncope.Hypotension
HypoglycemiaHypoglycemiaHypoglycemia is an emergency condition defined as a serum glucose level ≤ 70 mg/dL (≤ 3.9 mmol/L) in diabetic patients. In nondiabetic patients, there is no specific or defined limit for normal serum glucose levels, and hypoglycemia is defined mainly by its clinical features. Hypoglycemia
HyperkalemiaHyperkalemiaHyperkalemia is defined as a serum potassium (K+) concentration >5.2 mEq/L. Homeostatic mechanisms maintain the serum K+ concentration between 3.5 and 5.2 mEq/L, despite marked variation in dietary intake. Hyperkalemia can be due to a variety of causes, which include transcellular shifts, tissue breakdown, inadequate renal excretion, and drugs. Hyperkalemia
FatigueFatigueThe state of weariness following a period of exertion, mental or physical, characterized by a decreased capacity for work and reduced efficiency to respond to stimuli.Fibromyalgia
Cardiac arrhythmias
DiabetesDiabetesDiabetes mellitus (DM) is a metabolic disease characterized by hyperglycemia and dysfunction of the regulation of glucose metabolism by insulin. Type 1 DM is diagnosed mostly in children and young adults as the result of autoimmune destruction of β cells in the pancreas and the resulting lack of insulin. Type 2 DM has a significant association with obesity and is characterized by insulin resistance.Diabetes Mellitus insipidus (↓ antidiuretic hormoneAntidiuretic hormoneAntidiuretic hormones released by the neurohypophysis of all vertebrates (structure varies with species) to regulate water balance and osmolarity. In general, vasopressin is a nonapeptide consisting of a six-amino-acid ring with a cysteine 1 to cysteine 6 disulfide bridge or an octapeptide containing a cystine. All mammals have arginine vasopressin except the pig with a lysine at position 8. Vasopressin, a vasoconstrictor, acts on the kidney collecting ducts to increase water reabsorption, increase blood volume and blood pressure.Hypernatremia, approximately 20% of patientsPatientsIndividuals participating in the health care system for the purpose of receiving therapeutic, diagnostic, or preventive procedures.Clinician–Patient Relationship):
PolydipsiaPolydipsiaExcessive thirst manifested by excessive fluid intake. It is characteristic of many diseases such as diabetes mellitus; diabetes insipidus; and nephrogenic diabetes insipidus. The condition may be psychogenic in origin.Arginine Vasopressin Disorders (Diabetes Insipidus)
PolyuriaPolyuriaUrination of a large volume of urine with an increase in urinary frequency, commonly seen in diabetes.Renal Potassium Regulation
Panhypopituitarism: leads to a combination of the above detailed clinical manifestations
Cognitive dysfunction and behavioral issues
Cognitive dysfunction and behavioral issues may be due to impact on the thalamusThalamusThe thalamus is a large, ovoid structure in the dorsal part of the diencephalon that is located between the cerebral cortex and midbrain. It consists of several interconnected nuclei of grey matter separated by the laminae of white matter. The thalamus is the main conductor of information that passes between the cerebral cortex and the periphery, spinal cord, or brain stem.Thalamus: Anatomy, hypothalamusHypothalamusThe hypothalamus is a collection of various nuclei within the diencephalon in the center of the brain. The hypothalamus plays a vital role in endocrine regulation as the primary regulator of the pituitary gland, and it is the major point of integration between the central nervous and endocrine systems.Hypothalamus, and frontalFrontalThe bone that forms the frontal aspect of the skull. Its flat part forms the forehead, articulating inferiorly with the nasal bone and the cheek bone on each side of the face.Skull: Anatomy lobes.
Hyperphagia
AnorexiaAnorexiaThe lack or loss of appetite accompanied by an aversion to food and the inability to eat. It is the defining characteristic of the disorder anorexia nervosa.Anorexia Nervosa
Short-term memoryMemoryComplex mental function having four distinct phases: (1) memorizing or learning, (2) retention, (3) recall, and (4) recognition. Clinically, it is usually subdivided into immediate, recent, and remote memory.Psychiatric Assessment deficits
Incontinence
Signs of increased intracranial pressureIntracranial PressureIdiopathic Intracranial Hypertension (ICPICPNormal intracranial pressure (ICP) is defined as < 15 mm Hg, whereas pathologically increased ICP is any pressure ≥ 20 mm Hg. Increased ICP may result from several etiologies, including trauma, intracranial hemorrhage, mass lesions, cerebral edema, increased CSF production, and decreased CSF absorption.Increased Intracranial Pressure (ICP))
PapilledemaPapilledemaSwelling of the optic disk, usually in association with increased intracranial pressure, characterized by hyperemia, blurring of the disk margins, microhemorrhages, blind spot enlargement, and engorgement of retinal veins. Chronic papilledema may cause optic atrophy and visual loss.Idiopathic Intracranial Hypertension
DiplopiaDiplopiaA visual symptom in which a single object is perceived by the visual cortex as two objects rather than one. Disorders associated with this condition include refractive errors; strabismus; oculomotor nerve diseases; trochlear nerve diseases; abducens nerve diseases; and diseases of the brain stem and occipital lobe.Myasthenia Gravis
NauseaNauseaAn unpleasant sensation in the stomach usually accompanied by the urge to vomit. Common causes are early pregnancy, sea and motion sickness, emotional stress, intense pain, food poisoning, and various enteroviruses.Antiemetics/vomitingVomitingThe forcible expulsion of the contents of the stomach through the mouth.Hypokalemia
HeadacheHeadacheThe symptom of pain in the cranial region. It may be an isolated benign occurrence or manifestation of a wide variety of headache disorders.Brain Abscess
Diagnosis of craniopharyngiomas should be a multidisciplinary process, including the involvement of specialists in endocrinology, neurology, neuro-ophthalmology, and neurosurgeryNeurosurgeryNeurosurgery is a specialized field focused on the surgical management of pathologies of the brain, spine, spinal cord, and peripheral nerves. General neurosurgery includes cases of trauma and emergencies. There are a number of specialized neurosurgical practices, including oncologic neurosurgery, spinal neurosurgery, and pediatric neurosurgery. Neurosurgery.
Imaging
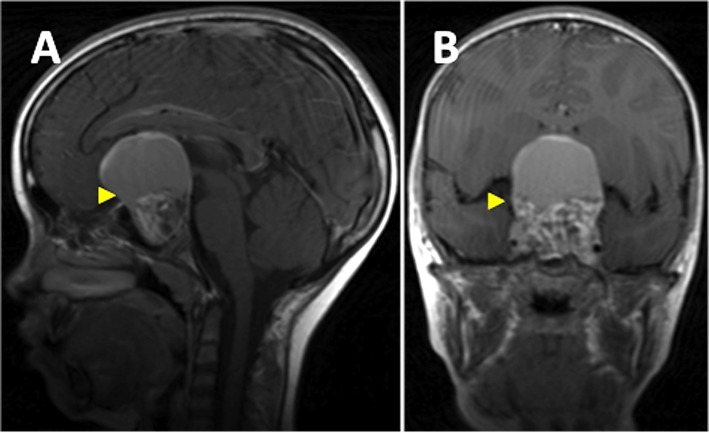
The main approach to the diagnosis of craniopharyngiomas is by using radiological imaging, specifically MRI and CT, which reveal a sellar/suprasellar calcified cyst (note: calcification is rare with papillary type). The tumors may have both solid and cysticCysticFibrocystic Change components.
MRI with and without contrast:
Considered the gold standard
Provides better information on the tumorTumorInflammation location and its association with the surrounding structures
CystsCystsAny fluid-filled closed cavity or sac that is lined by an epithelium. Cysts can be of normal, abnormal, non-neoplastic, or neoplastic tissues.Fibrocystic Change can have a variableVariableVariables represent information about something that can change. The design of the measurement scales, or of the methods for obtaining information, will determine the data gathered and the characteristics of that data. As a result, a variable can be qualitative or quantitative, and may be further classified into subgroups.Types of Variables appearance depending on the protein, blood, and/or cholesterolCholesterolThe principal sterol of all higher animals, distributed in body tissues, especially the brain and spinal cord, and in animal fats and oils.Cholesterol Metabolism content.
The best test to detect calcifications, as they are seen vividly on CT
Tumors enhance with contrast.
CystsCystsAny fluid-filled closed cavity or sac that is lined by an epithelium. Cysts can be of normal, abnormal, non-neoplastic, or neoplastic tissues.Fibrocystic Change present as hypodense lesions (resemble the fluid density of CSF).
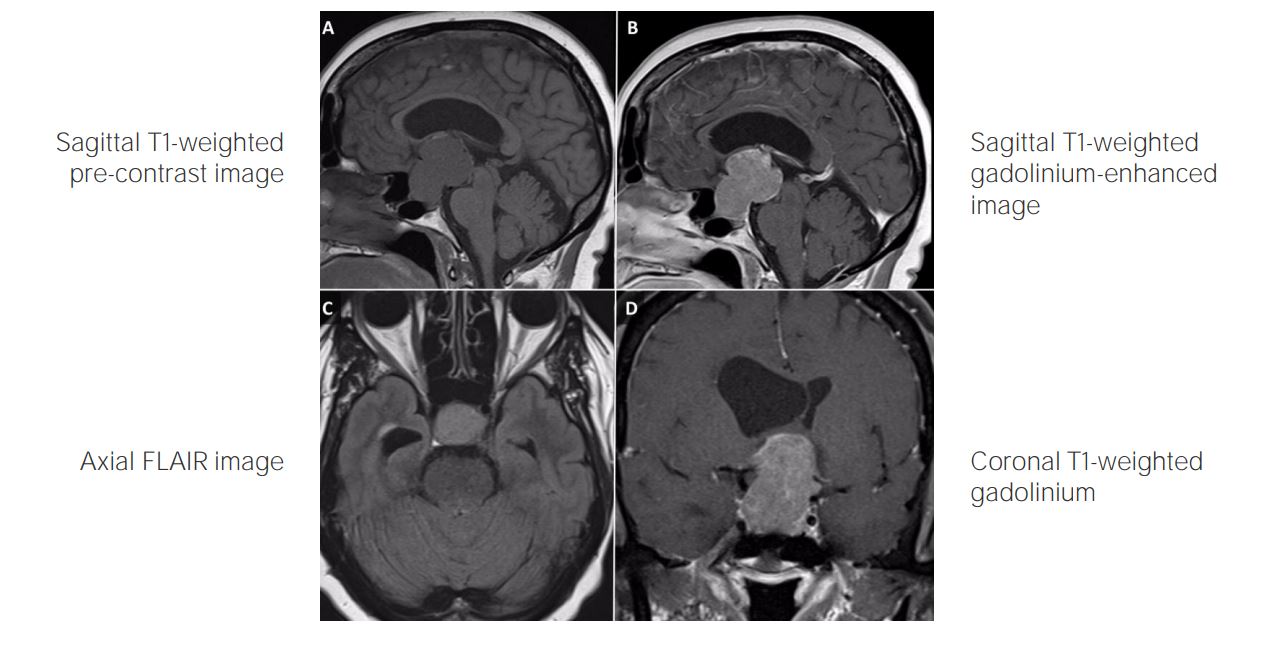
T1-weighted MRI of brain with contrast (sagittal view (panel A), coronal view (panel B)) shows a large, partly cystic, partly solid craniopharyngioma (arrowhead) with suprasellar extension elevating the optic chiasm and 3rd ventricle and causing sellar expansion.
Image: “Spontaneous Resolution of Radiotherapy-induced Craniopharyngioma Cyst” by Teo M, Cowie F, Fivey P, St George J. License: CC BY 3.0
Histology
Adamantinomatous type:
Reticular epithelial masses are observed.
Compactly arranged squamous cells
Palisading basal layer of small cells
A stellate reticular zone enclosed by the basal layer of cells
“Wet keratinKeratinA class of fibrous proteins or scleroproteins that represents the principal constituent of epidermis; hair; nails; horny tissues, and the organic matrix of tooth enamel. Two major conformational groups have been characterized, alpha-keratin, whose peptide backbone forms a coiled-coil alpha helical structure consisting of type I keratin and a type II keratin, and beta-keratin, whose backbone forms a zigzag or pleated sheet structure. Alpha-keratins have been classified into at least 20 subtypes. In addition multiple isoforms of subtypes have been found which may be due to gene duplication.Seborrheic Keratosis”: nodules of keratinKeratinA class of fibrous proteins or scleroproteins that represents the principal constituent of epidermis; hair; nails; horny tissues, and the organic matrix of tooth enamel. Two major conformational groups have been characterized, alpha-keratin, whose peptide backbone forms a coiled-coil alpha helical structure consisting of type I keratin and a type II keratin, and beta-keratin, whose backbone forms a zigzag or pleated sheet structure. Alpha-keratins have been classified into at least 20 subtypes. In addition multiple isoforms of subtypes have been found which may be due to gene duplication.Seborrheic Keratosis that appear plump (aggregated wet keratinKeratinA class of fibrous proteins or scleroproteins that represents the principal constituent of epidermis; hair; nails; horny tissues, and the organic matrix of tooth enamel. Two major conformational groups have been characterized, alpha-keratin, whose peptide backbone forms a coiled-coil alpha helical structure consisting of type I keratin and a type II keratin, and beta-keratin, whose backbone forms a zigzag or pleated sheet structure. Alpha-keratins have been classified into at least 20 subtypes. In addition multiple isoforms of subtypes have been found which may be due to gene duplication.Seborrheic Keratosis is what calcifies)
Papillary type:
Islands of squamous metaplasiaMetaplasiaA condition in which there is a change of one adult cell type to another similar adult cell type.Cellular Adaptation and fibrovascular tissue seen in connective tissueConnective tissueConnective tissues originate from embryonic mesenchyme and are present throughout the body except inside the brain and spinal cord. The main function of connective tissues is to provide structural support to organs. Connective tissues consist of cells and an extracellular matrix.Connective Tissue: Histology stroma
No keratinKeratinA class of fibrous proteins or scleroproteins that represents the principal constituent of epidermis; hair; nails; horny tissues, and the organic matrix of tooth enamel. Two major conformational groups have been characterized, alpha-keratin, whose peptide backbone forms a coiled-coil alpha helical structure consisting of type I keratin and a type II keratin, and beta-keratin, whose backbone forms a zigzag or pleated sheet structure. Alpha-keratins have been classified into at least 20 subtypes. In addition multiple isoforms of subtypes have been found which may be due to gene duplication.Seborrheic Keratosis nodules
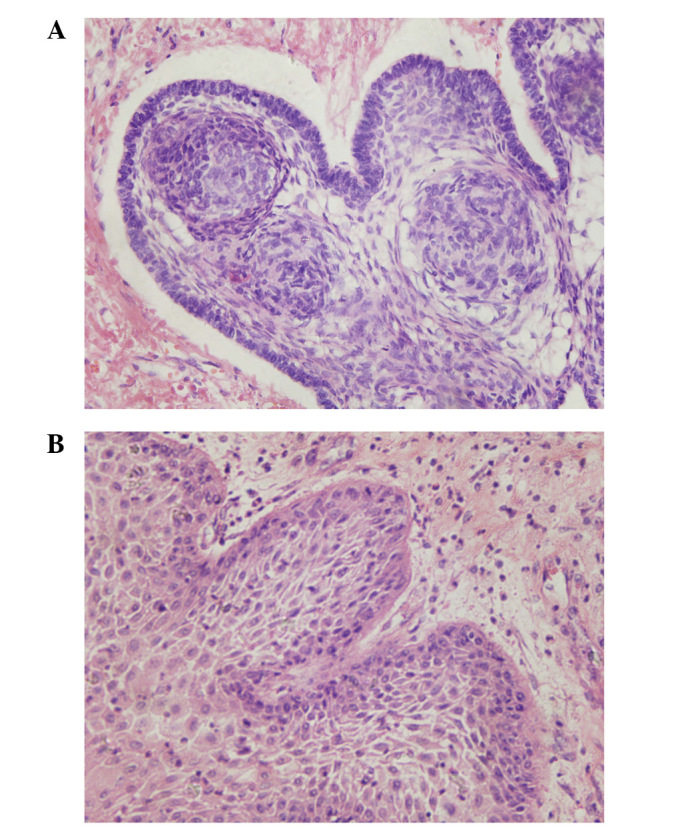
Adamantinomatous (panel A) and squamous-papillary (panel B) craniopharyngioma section images captured using light microscopy (stain, hematoxylin and eosin; magnification, ×400)
Image: “Craniopharyngioma: Survivin expression and ultrastructure” by Zhu J, You C. License: CC BY 3.0
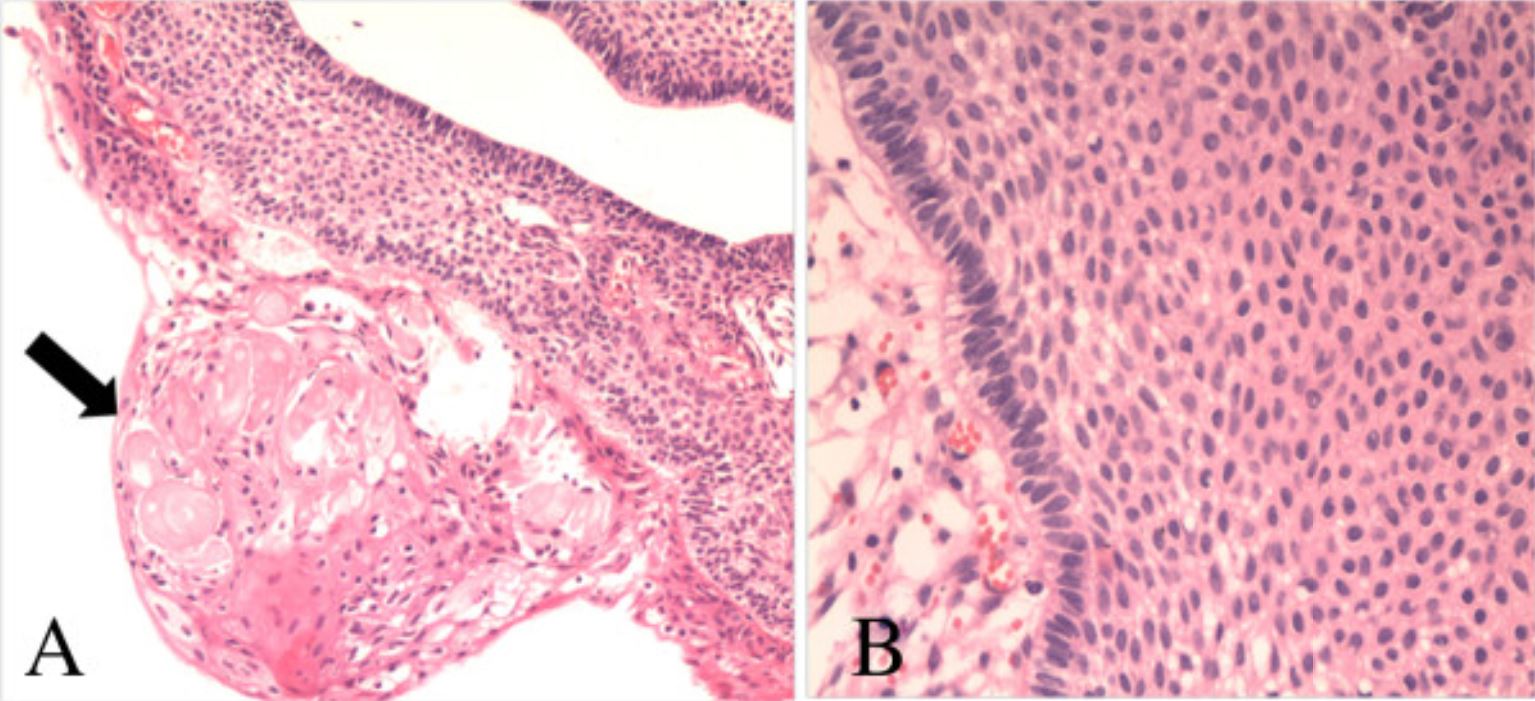
Photomicrographs of the pathological specimen showing features of an adamantinomatous craniopharyngioma: A: wet keratin (arrow, hematoxylin and eosin (H&E); magnification, ×100) B: multiple layers of squamous epithelium (H&E; magnification, ×200)
Image: “Collision tumors of the sella: coexistence of pituitary adenoma and craniopharyngioma in the sellar region” by Jin G, Hao S, Xie J, Mi R, Liu F. License: CC BY 2.0
Additional workup
The following should be completed if not done at initial presentation:
Full neuro exam
Ophthalmology assessment
Endocrine workup:
TSH levels
GH levels
Gonadotropin levels:
Follicle stimulating hormone (FSHFSHA major gonadotropin secreted by the adenohypophysis. Follicle-stimulating hormone stimulates gametogenesis and the supporting cells such as the ovarian granulosa cells, the testicular sertoli cells, and leydig cells. Fsh consists of two noncovalently linked subunits, alpha and beta. Within a species, the alpha subunit is common in the three pituitary glycoprotein hormones (TSH, LH, and FSH), but the beta subunit is unique and confers its biological specificity.Menstrual Cycle)
Luteinizing hormone (LHLHA major gonadotropin secreted by the adenohypophysis. Luteinizing hormone regulates steroid production by the interstitial cells of the testis and the ovary. The preovulatory luteinizing hormone surge in females induces ovulation, and subsequent luteinization of the follicle. Luteinizing hormone consists of two noncovalently linked subunits, alpha and beta. Within a species, the alpha subunit is common in the three pituitary glycoprotein hormones (TSH, LH, and FSH), but the beta subunit is unique and confers its biological specificity.Menstrual Cycle)
ProlactinProlactinA lactogenic hormone secreted by the adenohypophysis. It is a polypeptide of approximately 23 kd. Besides its major action on lactation, in some species prolactin exerts effects on reproduction, maternal behavior, fat metabolism, immunomodulation and osmoregulation.Breasts: Anatomy
Serum electrolytesElectrolytesElectrolytes are mineral salts that dissolve in water and dissociate into charged particles called ions, which can be either be positively (cations) or negatively (anions) charged. Electrolytes are distributed in the extracellular and intracellular compartments in different concentrations. Electrolytes are essential for various basic life-sustaining functions.Electrolytes
Considered more often to ensure a good qualityQualityActivities and programs intended to assure or improve the quality of care in either a defined medical setting or a program. The concept includes the assessment or evaluation of the quality of care; identification of problems or shortcomings in the delivery of care; designing activities to overcome these deficiencies; and follow-up monitoring to ensure effectiveness of corrective steps.Quality Measurement and Improvement of life postoperatively
RadiationRadiationEmission or propagation of acoustic waves (sound), electromagnetic energy waves (such as light; radio waves; gamma rays; or x-rays), or a stream of subatomic particles (such as electrons; neutrons; protons; or alpha particles).Osteosarcoma therapy options:
Stereotactic radiosurgery
Intensity-modulated radiationRadiationEmission or propagation of acoustic waves (sound), electromagnetic energy waves (such as light; radio waves; gamma rays; or x-rays), or a stream of subatomic particles (such as electrons; neutrons; protons; or alpha particles).Osteosarcoma therapy
Proton beam radiationRadiationEmission or propagation of acoustic waves (sound), electromagnetic energy waves (such as light; radio waves; gamma rays; or x-rays), or a stream of subatomic particles (such as electrons; neutrons; protons; or alpha particles).Osteosarcoma therapy
Cyst management
While surgical management is typically required, there are additional options available to reduce cyst size/massMassThree-dimensional lesion that occupies a space within the breastImaging of the Breast effect, including:
Intracavitary chemotherapyChemotherapyOsteosarcoma (can leak from a cyst and cause severe neurotoxicity)
Complications
Craniopharyngiomas can cause several complications:
Endocrine
Neurologic
Visual
Vascular
PatientsPatientsIndividuals participating in the health care system for the purpose of receiving therapeutic, diagnostic, or preventive procedures.Clinician–Patient Relationship should be managed by multidisciplinary teams to address issues specific to each patient.
PrognosisPrognosisA prediction of the probable outcome of a disease based on a individual’s condition and the usual course of the disease as seen in similar situations.Non-Hodgkin Lymphomas
The prognosisPrognosisA prediction of the probable outcome of a disease based on a individual’s condition and the usual course of the disease as seen in similar situations.Non-Hodgkin Lymphomas of craniopharyngiomas depends on the recurrence of the tumorTumorInflammation.
Frequent recurrence is usually associated with a poor prognosisPrognosisA prediction of the probable outcome of a disease based on a individual’s condition and the usual course of the disease as seen in similar situations.Non-Hodgkin Lymphomas.
Survival rates:
Approximately 90% survival rate at 2 years
Approximately 85% survival rate at 5 years
5-year survival based on age:
93% in patientsPatientsIndividuals participating in the health care system for the purpose of receiving therapeutic, diagnostic, or preventive procedures.Clinician–Patient Relationship 1‒14 years of age
88% in adolescents and young adults
78% in patientsPatientsIndividuals participating in the health care system for the purpose of receiving therapeutic, diagnostic, or preventive procedures.Clinician–Patient Relationship > 40 years of age
The conditions listed below should be considered in the differential diagnosis for craniopharyngiomas. Most cases will present similarly and can be differentiated based on differences in imaging and histology.
PituitaryPituitaryA small, unpaired gland situated in the sella turcica. It is connected to the hypothalamus by a short stalk which is called the infundibulum.Hormones: Overview and Types adenomas: benignBenignFibroadenoma tumors within the pituitaryPituitaryA small, unpaired gland situated in the sella turcica. It is connected to the hypothalamus by a short stalk which is called the infundibulum.Hormones: Overview and Types gland, many of which are hormonally active. The most common type is a prolactinomaProlactinomaA pituitary adenoma which secretes prolactin, leading to hyperprolactinemia. Clinical manifestations include amenorrhea; galactorrhea; impotence; headache; visual disturbances; and cerebrospinal fluid rhinorrhea.Hyperprolactinemia, although they may secrete any of the hormonesHormonesHormones are messenger molecules that are synthesized in one part of the body and move through the bloodstream to exert specific regulatory effects on another part of the body. Hormones play critical roles in coordinating cellular activities throughout the body in response to the constant changes in both the internal and external environments. Hormones: Overview and Types produced by the pituitaryPituitaryA small, unpaired gland situated in the sella turcica. It is connected to the hypothalamus by a short stalk which is called the infundibulum.Hormones: Overview and Types gland. The clinical presentation may include headacheHeadacheThe symptom of pain in the cranial region. It may be an isolated benign occurrence or manifestation of a wide variety of headache disorders.Brain Abscess, fatigueFatigueThe state of weariness following a period of exertion, mental or physical, characterized by a decreased capacity for work and reduced efficiency to respond to stimuli.Fibromyalgia, bitemporal hemianopiaHemianopiaTransient Ischemic Attack (TIA) and/or diplopiaDiplopiaA visual symptom in which a single object is perceived by the visual cortex as two objects rather than one. Disorders associated with this condition include refractive errors; strabismus; oculomotor nerve diseases; trochlear nerve diseases; abducens nerve diseases; and diseases of the brain stem and occipital lobe.Myasthenia Gravis, and findings associated with elevated hormone levels. Like craniopharyngioma, pituitaryPituitaryA small, unpaired gland situated in the sella turcica. It is connected to the hypothalamus by a short stalk which is called the infundibulum.Hormones: Overview and Types adenomas are located within the sella, but only rarely contain cystsCystsAny fluid-filled closed cavity or sac that is lined by an epithelium. Cysts can be of normal, abnormal, non-neoplastic, or neoplastic tissues.Fibrocystic Change or calcifications.
Glioblastoma multiformeGlioblastoma multiformeGlioblastoma multiforme is a high-grade astrocytoma, an aggressive brain tumor arising from astrocytes, with an unknown cause and a poorly understood link to risk factors. There are two main types: primary, a more aggressive form seen more commonly in older patients, and secondary, developing from lower-grade astrocytomas and seen more commonly in younger patients.Glioblastoma Multiforme: a rapidly progressive WHO grade IV astrocytomaGrade IV AstrocytomaAstrocytoma that arises from astrocytesAstrocytesA class of large neuroglial (macroglial) cells in the central nervous system – the largest and most numerous neuroglial cells in the brain and spinal cord. Astrocytes (from ‘star’ cells) are irregularly shaped with many long processes, including those with ‘end feet’ which form the glial (limiting) membrane and directly and indirectly contribute to the blood-brain barrier. They regulate the extracellular ionic and chemical environment, and ‘reactive astrocytes’ (along with microglia) respond to injury.Nervous System: Histology (glial cellsGlial CellsThe non-neuronal cells of the nervous system. They not only provide physical support, but also respond to injury, regulate the ionic and chemical composition of the extracellular milieu, participate in the blood-brain barrier and blood-retinal barrier, form the myelin insulation of nervous pathways, guide neuronal migration during development, and exchange metabolites with neurons. Neuroglia have high-affinity transmitter uptake systems, voltage-dependent and transmitter-gated ion channels, and can release transmitters, but their role in signaling (as in many other functions) is unclear.Nervous System: Histology in the brainBrainThe part of central nervous system that is contained within the skull (cranium). Arising from the neural tube, the embryonic brain is comprised of three major parts including prosencephalon (the forebrain); mesencephalon (the midbrain); and rhombencephalon (the hindbrain). The developed brain consists of cerebrum; cerebellum; and other structures in the brain stem.Nervous System: Anatomy, Structure, and Classification) and clinically presents as headacheHeadacheThe symptom of pain in the cranial region. It may be an isolated benign occurrence or manifestation of a wide variety of headache disorders.Brain Abscess, nauseaNauseaAn unpleasant sensation in the stomach usually accompanied by the urge to vomit. Common causes are early pregnancy, sea and motion sickness, emotional stress, intense pain, food poisoning, and various enteroviruses.Antiemetics, drowsiness, blurred visionBlurred VisionRetinal Detachment, personality changes, and seizuresSeizuresA seizure is abnormal electrical activity of the neurons in the cerebral cortex that can manifest in numerous ways depending on the region of the brain affected. Seizures consist of a sudden imbalance that occurs between the excitatory and inhibitory signals in cortical neurons, creating a net excitation. The 2 major classes of seizures are focal and generalized. Seizures. Imaging, clinical presentation, and biopsyBiopsyRemoval and pathologic examination of specimens from the living body.Ewing Sarcoma are the mainstays for diagnosis. Management includes radiotherapy, chemotherapyChemotherapyOsteosarcoma, and surgical excision. PrognosisPrognosisA prediction of the probable outcome of a disease based on a individual’s condition and the usual course of the disease as seen in similar situations.Non-Hodgkin Lymphomas is poor even with treatment. Unlike hemangioblastomaHemangioblastomaHemangioblastomas are vascular neoplasms of the CNS. Hemangioblastomas are rare and are often associated with von Hippel-Lindau disease (VHL). The most common presentation is a headache and, depending on the size and location of the tumor, patients may present with sensory deficits and motor weakness.Hemangioblastoma, glioblastoma multiformeGlioblastoma multiformeGlioblastoma multiforme is a high-grade astrocytoma, an aggressive brain tumor arising from astrocytes, with an unknown cause and a poorly understood link to risk factors. There are two main types: primary, a more aggressive form seen more commonly in older patients, and secondary, developing from lower-grade astrocytomas and seen more commonly in younger patients.Glioblastoma Multiforme is not associated with von Hippel-Lindau (VHL) disease.
HemangioblastomaHemangioblastomaHemangioblastomas are vascular neoplasms of the CNS. Hemangioblastomas are rare and are often associated with von Hippel-Lindau disease (VHL). The most common presentation is a headache and, depending on the size and location of the tumor, patients may present with sensory deficits and motor weakness.Hemangioblastoma: a rare vascular tumorTumorInflammation of the CNS, often associated with VHL diseaseVHL diseaseVon Hippel-Lindau (VHL) disease is an autosomal dominant genetic condition resulting from a deletion or mutation in the VHL gene. Individuals diagnosed with VHL disease have tumors and cysts in various parts of their bodies and may present with hemangioblastomas, renal cell carcinoma (RCC), pheochromocytoma, endolymphatic sac tumors of the middle ear, pancreatic tumors, and papillary cystadenomas of the epididymis or the broad ligament. Von Hippel-Lindau Disease, which usually affects the brainBrainThe part of central nervous system that is contained within the skull (cranium). Arising from the neural tube, the embryonic brain is comprised of three major parts including prosencephalon (the forebrain); mesencephalon (the midbrain); and rhombencephalon (the hindbrain). The developed brain consists of cerebrum; cerebellum; and other structures in the brain stem.Nervous System: Anatomy, Structure, and Classification but can also involve the spinal cordSpinal cordThe spinal cord is the major conduction pathway connecting the brain to the body; it is part of the CNS. In cross section, the spinal cord is divided into an H-shaped area of gray matter (consisting of synapsing neuronal cell bodies) and a surrounding area of white matter (consisting of ascending and descending tracts of myelinated axons). Spinal Cord: Anatomy or retinaRetinaThe ten-layered nervous tissue membrane of the eye. It is continuous with the optic nerve and receives images of external objects and transmits visual impulses to the brain. Its outer surface is in contact with the choroid and the inner surface with the vitreous body. The outermost layer is pigmented, whereas the inner nine layers are transparent.Eye: Anatomy. Presenting symptoms include headacheHeadacheThe symptom of pain in the cranial region. It may be an isolated benign occurrence or manifestation of a wide variety of headache disorders.Brain Abscess and neurological deficits based on the location of the tumorTumorInflammation. While hemangioblastomasHemangioblastomasA benign tumor of the nervous system that may occur sporadically or in association with von Hippel-Lindau disease. It accounts for approximately 2% of intracranial tumors, arising most frequently in the cerebellar hemispheres and vermis. Histologically, the tumors are composed of multiple capillary and sinusoidal channels lined with endothelial cells and clusters of lipid-laden pseudoxanthoma cells. Usually solitary, these tumors can be multiple and may also occur in the brain stem, spinal cord, retina, and supratentorial compartment. Cerebellar hemangioblastomas usually present in the third decade with intracranial hypertension, and ataxia.Von Hippel-Lindau Disease may have both solid and cysticCysticFibrocystic Change components, they tend to be located in the cerebellumCerebellumThe cerebellum, Latin for “little brain,” is located in the posterior cranial fossa, dorsal to the pons and midbrain, and its principal role is in the coordination of movements. The cerebellum consists of 3 lobes on either side of its 2 hemispheres and is connected in the middle by the vermis. Cerebellum: Anatomy or spinal cordSpinal cordThe spinal cord is the major conduction pathway connecting the brain to the body; it is part of the CNS. In cross section, the spinal cord is divided into an H-shaped area of gray matter (consisting of synapsing neuronal cell bodies) and a surrounding area of white matter (consisting of ascending and descending tracts of myelinated axons). Spinal Cord: Anatomy and do not generally calcify.
MeningiomaMeningiomaMeningiomas are slow-growing tumors that arise from the meninges of the brain and spinal cord. The vast majority are benign. These tumors commonly occur in individuals with a history of high doses of skull radiation, head trauma, and neurofibromatosis 2. Meningioma: a tumorTumorInflammation arising from the meningesMeningesThe brain and the spinal cord are enveloped by 3 overlapping layers of connective tissue called the meninges. The layers are, from the most external layer to the most internal layer, the dura mater, arachnoid mater, and pia mater. Between these layers are 3 potential spaces called the epidural, subdural, and subarachnoid spaces. Meninges: Anatomy of the brainBrainThe part of central nervous system that is contained within the skull (cranium). Arising from the neural tube, the embryonic brain is comprised of three major parts including prosencephalon (the forebrain); mesencephalon (the midbrain); and rhombencephalon (the hindbrain). The developed brain consists of cerebrum; cerebellum; and other structures in the brain stem.Nervous System: Anatomy, Structure, and Classification and spinal cordSpinal cordThe spinal cord is the major conduction pathway connecting the brain to the body; it is part of the CNS. In cross section, the spinal cord is divided into an H-shaped area of gray matter (consisting of synapsing neuronal cell bodies) and a surrounding area of white matter (consisting of ascending and descending tracts of myelinated axons). Spinal Cord: Anatomy. Meningiomas are commonly asymptomatic, but can present with headacheHeadacheThe symptom of pain in the cranial region. It may be an isolated benign occurrence or manifestation of a wide variety of headache disorders.Brain Abscess, seizuresSeizuresA seizure is abnormal electrical activity of the neurons in the cerebral cortex that can manifest in numerous ways depending on the region of the brain affected. Seizures consist of a sudden imbalance that occurs between the excitatory and inhibitory signals in cortical neurons, creating a net excitation. The 2 major classes of seizures are focal and generalized. Seizures, and visual disturbances. Meningiomas are diagnosed using MRI and a biopsyBiopsyRemoval and pathologic examination of specimens from the living body.Ewing Sarcoma. Asymptomatic cases are typically observed while symptomatic patientsPatientsIndividuals participating in the health care system for the purpose of receiving therapeutic, diagnostic, or preventive procedures.Clinician–Patient Relationship are treated surgically or with radiationRadiationEmission or propagation of acoustic waves (sound), electromagnetic energy waves (such as light; radio waves; gamma rays; or x-rays), or a stream of subatomic particles (such as electrons; neutrons; protons; or alpha particles).Osteosarcoma. Unlike craniopharyngiomas, meningiomas are always near the meningesMeningesThe brain and the spinal cord are enveloped by 3 overlapping layers of connective tissue called the meninges. The layers are, from the most external layer to the most internal layer, the dura mater, arachnoid mater, and pia mater. Between these layers are 3 potential spaces called the epidural, subdural, and subarachnoid spaces. Meninges: Anatomy and often have imaging findings of dural attachment (e.g., dural tail signDural tail signMarginal dural thickening that tapers peripherallyMeningioma).
Kassam, A.B., et al. (2008). Expanded endonasal approach, a fully endoscopic transnasal approach for the resection of midline suprasellar craniopharyngiomas: A new classification based on the infundibulum. J Neurosurg. 108(4), 715–728. https://pubmed.ncbi.nlm.nih.gov/18377251/
Create your free account or log in to continue reading!