Pheochromocytoma is a catecholamine-secreting tumor derived from chromaffin cells. The term pheochromocytoma is used for tumors arising from the adrenal medulla, while paraganglioma is used for tumors arising from extra-adrenal sympathetic or parasympathetic ganglia. Symptoms are associated with excessive catecholamine production and commonly include hypertension Hypertension Hypertension, or high blood pressure, is a common disease that manifests as elevated systemic arterial pressures. Hypertension is most often asymptomatic and is found incidentally as part of a routine physical examination or during triage for an unrelated medical encounter. Hypertension, tachycardia Tachycardia Abnormally rapid heartbeat, usually with a heart rate above 100 beats per minute for adults. Tachycardia accompanied by disturbance in the cardiac depolarization (cardiac arrhythmia) is called tachyarrhythmia. Sepsis in Children, headache Headache The symptom of pain in the cranial region. It may be an isolated benign occurrence or manifestation of a wide variety of headache disorders. Brain Abscess, and sweating. Pheochromocytomas are frequently familial and associated with genetic syndromes. Approximately 90% of pheochromocytomas are benign Benign Fibroadenoma and surgical resection is the only curative treatment. Prognosis Prognosis A prediction of the probable outcome of a disease based on a individual's condition and the usual course of the disease as seen in similar situations. Non-Hodgkin Lymphomas is generally good for benign Benign Fibroadenoma, sporadic Sporadic Selective IgA Deficiency tumors that have been completely resected. Familial forms are associated with higher recurrence rates and malignant potential.
Last updated: Dec 15, 2025
Pheochromocytoma is a catecholamine-secreting tumor Tumor Inflammation derived from chromaffin cells Chromaffin cells Cells that store epinephrine secretory vesicles. During times of stress, the nervous system signals the vesicles to secrete their hormonal content. Their name derives from their ability to stain a brownish color with chromic salts. Characteristically, they are located in the adrenal medulla and paraganglia of the sympathetic nervous system. Adrenal Hormones.
The “rule of 10s” for pheochromocytoma characteristics:
To recall the most common clinical features of pheochromocytoma or episodic hyperadrenergic spells, remember the “5 Ps“:
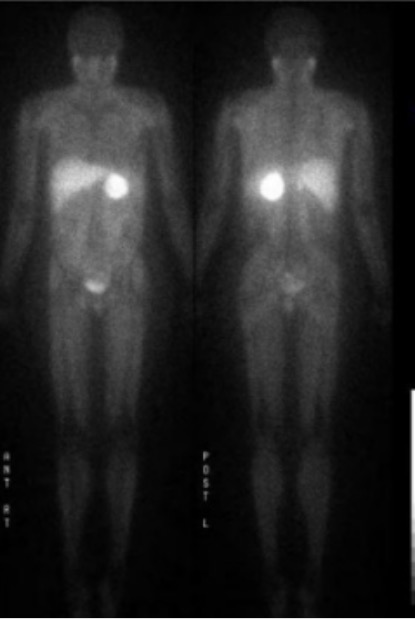
Intense uptake by an adrenal mass (pheochromocytoma) as shown on a 123I-MIBG scan
Image: “123I-MIBG scan” by Endocrine Unit, Department of Medicine, University of Rochester, Rochester, NY, 14642, USA. License: CC BY 2.0, cropped by Lecturio.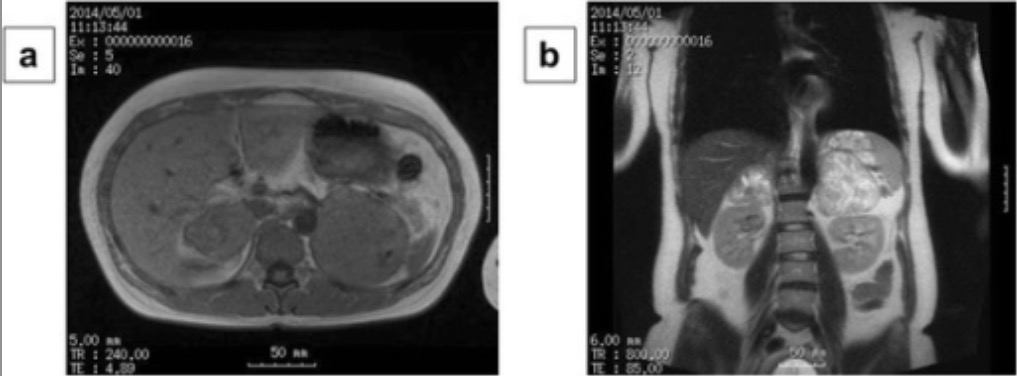
Bilateral adrenal pheochromocytoma with central necrosis as shown on MRI
Image: “MRI findings” by Department of Surgical Oncology, Osaka City University Graduate School of Medicine, 1-4-3 Asahi-machi, Abeno-ku, Osaka, Japan. License: CC BY 4.0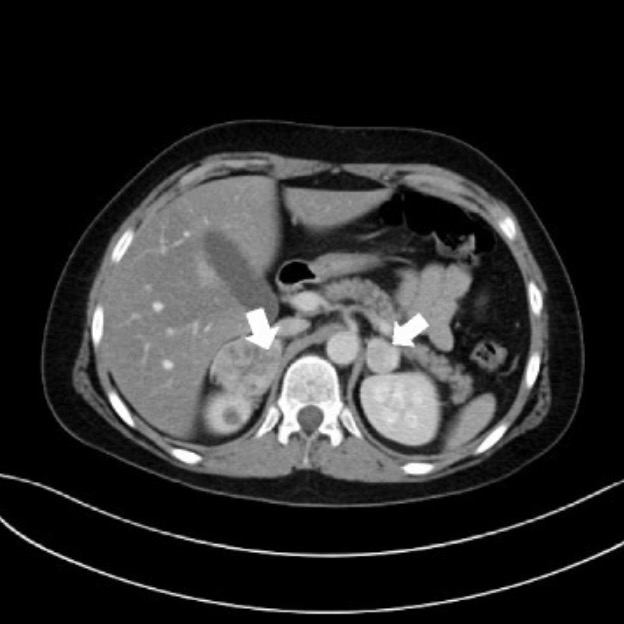
Bilateral adrenal pheochromocytoma in a patient with VHL disease
Image: “Bilateral nodular lesions” by Department of Paediatric Endocrinology and Diabetology, Medical University in Lublin, Poland, ul. Prof. A. Gebali 6, 20-093, Lublin, Poland. License: CC BY 4.0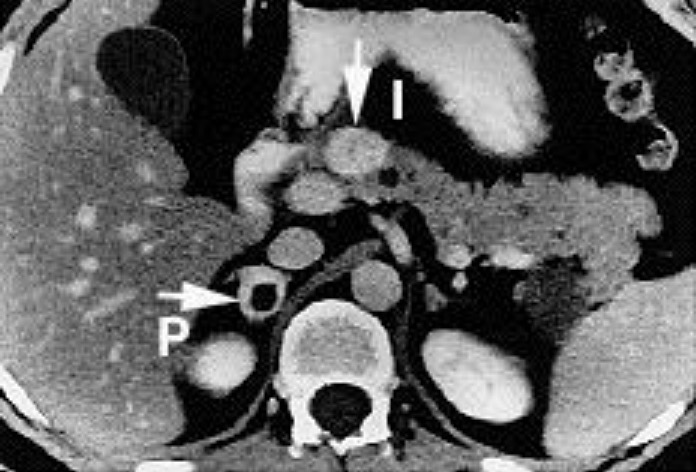
Computed tomography of a patient presenting simultaneous pheochromocytoma and islet cell tumor of the pancreas:
Note the pheochromocytoma has necrotic center.
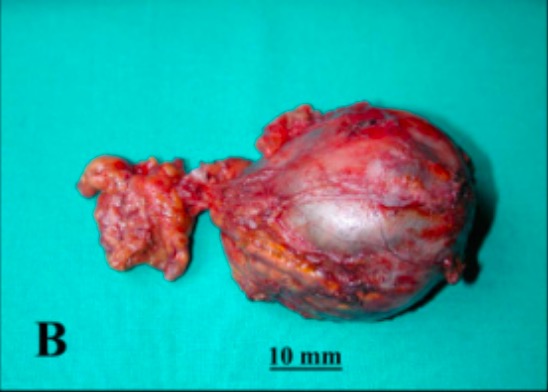
Pheochromocytoma of left adrenal gland (post-resection)
Image: “Pre-surgical MR and macroscopic anatomy images” by Non-invasive Cardiac Imaging Laboratory, CMR Unit, Department of Cardiology and Cardiovascular Surgery, Niguarda Ca’Granda Hospital, Milan, Italy. License: CC BY 2.0, cropped by Lecturio.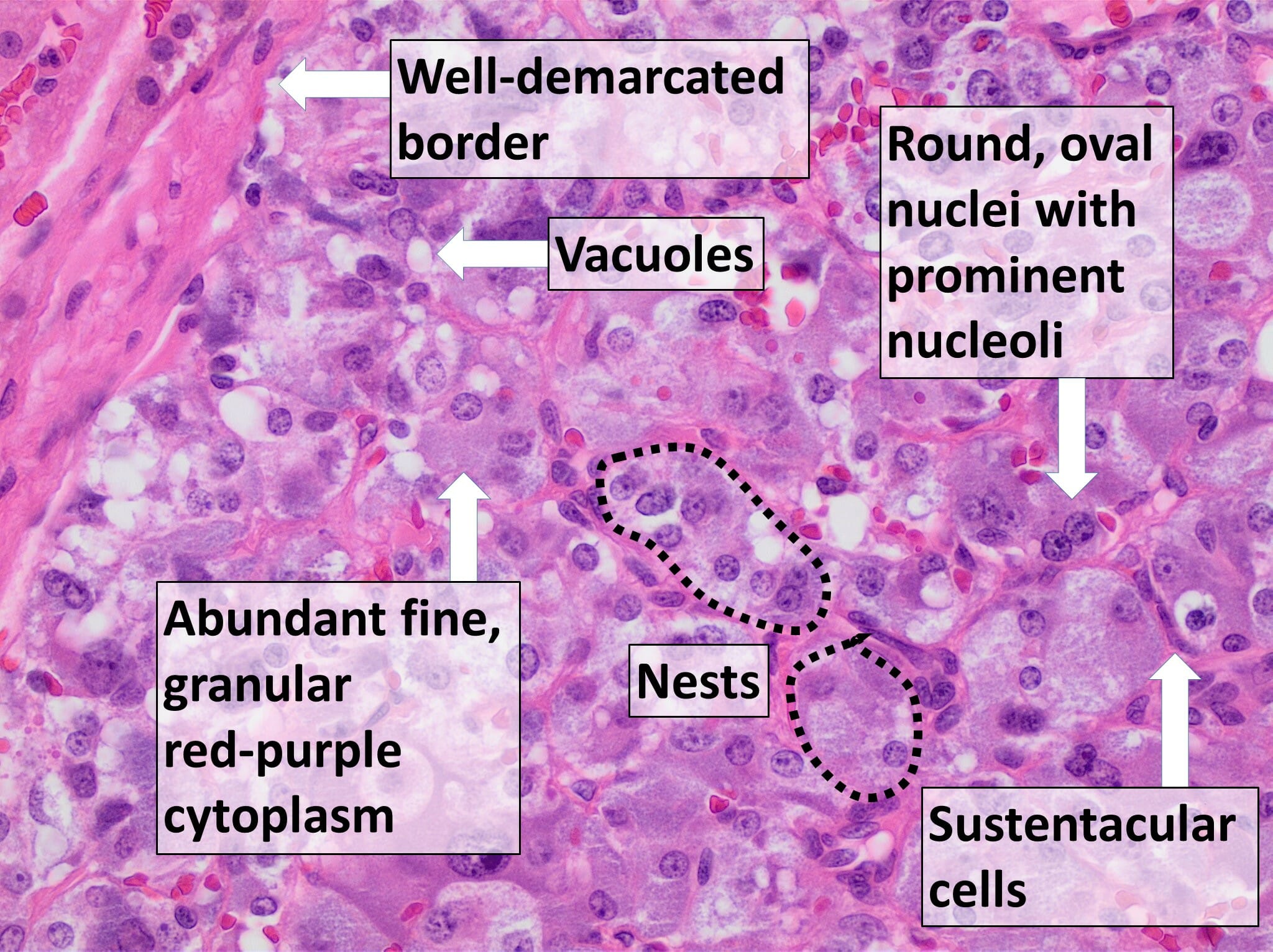
Histological specimen of pheochromocytoma:
The diagnosis of pheochromocytoma is confirmed by microscopic evaluation of the resected tumor after surgical resection. The important features observed include nests or trabecular arrangement of cells, extensive vacuolation of cells, large and polygonal sustentacular cells, granular red-purple cytoplasm, round or oval nuclei, and prominent nucleoli.