IdiopathicIdiopathicDermatomyositis pulmonary fibrosisFibrosisAny pathological condition where fibrous connective tissue invades any organ, usually as a consequence of inflammation or other injury.Bronchiolitis Obliterans is a specific entity of the major idiopathicIdiopathicDermatomyositis interstitial pneumoniaPneumoniaPneumonia or pulmonary inflammation is an acute or chronic inflammation of lung tissue. Causes include infection with bacteria, viruses, or fungi. In more rare cases, pneumonia can also be caused through toxic triggers through inhalation of toxic substances, immunological processes, or in the course of radiotherapy.Pneumonia classification of interstitial lung diseasesInterstitial Lung DiseasesInterstitial lung diseases are a heterogeneous group of disorders characterized by the inflammation and fibrosis of lung parenchyma, especially the pulmonary connective tissue in the alveolar walls. It may be idiopathic (e.g., idiopathic pulmonary fibrosis) or secondary to connective tissue diseases, medications, malignancies, occupational exposure, or allergens.Interstitial Lung Diseases. As implied by the name, the exact causes are poorly understood. PatientsPatientsIndividuals participating in the health care system for the purpose of receiving therapeutic, diagnostic, or preventive procedures.Clinician–Patient Relationship often present in the moderate to advanced stage with progressive dyspneaDyspneaDyspnea is the subjective sensation of breathing discomfort. Dyspnea is a normal manifestation of heavy physical or psychological exertion, but also may be caused by underlying conditions (both pulmonary and extrapulmonary). Dyspnea and nonproductive cough. The diagnosis is made by characteristic imaging findings, pulmonary function testingPulmonary Function TestingPulmonary Function Tests that indicates restrictive lung disease, and (if necessary) lung biopsyLung BiopsyAntineutrophil Cytoplasmic Antibody (ANCA)-Associated Vasculitis. Options are limited for therapies to slow progression. Lung transplantationLung transplantationThe transference of either one or both of the lungs from one human or animal to another.Organ Transplantation is the only curative intervention if the patient is a candidate for one.
IdiopathicIdiopathicDermatomyositis pulmonary fibrosisFibrosisAny pathological condition where fibrous connective tissue invades any organ, usually as a consequence of inflammation or other injury.Bronchiolitis Obliterans (IPF) is the most common type of interstitial lung disease (ILDILDInterstitial lung diseases are a heterogeneous group of disorders characterized by the inflammation and fibrosis of lung parenchyma, especially the pulmonary connective tissue in the alveolar walls. It may be idiopathic (e.g., idiopathic pulmonary fibrosis) or secondary to connective tissue diseases, medications, malignancies, occupational exposure, or allergens.Interstitial Lung Diseases) and is characterized by chronic, progressive, irreversible fibrosisFibrosisAny pathological condition where fibrous connective tissue invades any organ, usually as a consequence of inflammation or other injury.Bronchiolitis Obliterans of the lung parenchyma.
Epidemiology
IPF has been difficult to study because of its rarity and evolution in diagnostic practices.
IncidenceIncidenceThe number of new cases of a given disease during a given period in a specified population. It also is used for the rate at which new events occur in a defined population. It is differentiated from prevalence, which refers to all cases in the population at a given time.Measures of Disease Frequency and prevalencePrevalenceThe total number of cases of a given disease in a specified population at a designated time. It is differentiated from incidence, which refers to the number of new cases in the population at a given time.Measures of Disease Frequency:
Uncommon
Higher in North America and Europe than in the rest of the world
Estimated prevalencePrevalenceThe total number of cases of a given disease in a specified population at a designated time. It is differentiated from incidence, which refers to the number of new cases in the population at a given time.Measures of Disease Frequency in the United States: 10–60 per 100,000
Likely underappreciated in terms of its occurrence and public health impact (e.g., health care costs and resource utilization)
Occurs primarily in elderly individuals (> 65 years)
The cause of IPF remains largely unclear. However, in the current hypothesisHypothesisA hypothesis is a preliminary answer to a research question (i.e., a “guess” about what the results will be). There are 2 types of hypotheses: the null hypothesis and the alternative hypothesis.Statistical Tests and Data Representation regarding the pathogenesis of IPF, the following exposures may lead to the initial alveolar epithelial injury:
Tobacco smoke (most common)
Environmental pollutants and dust
Viral infectionsInfectionsInvasion of the host organism by microorganisms or their toxins or by parasites that can cause pathological conditions or diseases.Chronic Granulomatous Disease
GERDGERDGastroesophageal reflux disease (GERD) occurs when the stomach acid frequently flows back into the esophagus. This backwash (acid reflux) can irritate the lining of the esophagus, causing symptoms such as retrosternal burning pain (heartburn). Gastroesophageal Reflux Disease (GERD)
Chronic microaspiration
Obstructive sleep apneaSleep apneaRepeated cessation of breathing for > 10 seconds during sleep and results in sleep interruption, fatigue, and daytime sleepiness.Obstructive Sleep Apnea
Pathophysiology
The underlying mechanism is poorly understood.
Environmental exposure and possible genetic predisposition → recurrent alveolar epithelial damage
Migration and activation of fibroblastsFibroblastsConnective tissue cells which secrete an extracellular matrix rich in collagen and other macromolecules.Sarcoidosis → myofibroblast transformationTransformationChange brought about to an organism’s genetic composition by unidirectional transfer (transfection; transduction, genetic; conjugation, genetic, etc.) and incorporation of foreign DNA into prokaryotic or eukaryotic cells by recombination of part or all of that DNA into the cell’s genome.Bacteriology
Failure of normal myofibroblast apoptosisApoptosisA regulated cell death mechanism characterized by distinctive morphologic changes in the nucleus and cytoplasm, including the endonucleolytic cleavage of genomic DNA, at regularly spaced, internucleosomal sites, I.e., DNA fragmentation. It is genetically-programmed and serves as a balance to mitosis in regulating the size of animal tissues and in mediating pathologic processes associated with tumor growth.Ischemic Cell Damage → exaggerated extracellular matrixExtracellular matrixA meshwork-like substance found within the extracellular space and in association with the basement membrane of the cell surface. It promotes cellular proliferation and provides a supporting structure to which cells or cell lysates in culture dishes adhere.Hypertrophic and Keloid Scars accumulation → lung parenchyma destruction and scarringScarringInflammation
Additional mediators of profibrotic changes include:
Transforming growth factor beta
Fibroblast growth factorFibroblast growth factorA family of small polypeptide growth factors that share several common features including a strong affinity for heparin, and a central barrel-shaped core region of 140 amino acids that is highly homologous between family members. Although originally studied as proteins that stimulate the growth of fibroblasts this distinction is no longer a requirement for membership in the fibroblast growth factor family.X-linked Hypophosphatemic Rickets 2
Platelet-derived growth factorPlatelet-derived growth factorMitogenic peptide growth hormone carried in the alpha-granules of platelets. It is released when platelets adhere to traumatized tissues. Connective tissue cells near the traumatized region respond by initiating the process of replication.Hypertrophic and Keloid Scars (PDGF)
Matrix metalloproteinase 7 (MMP-7)
Consequence: ↓ gas exchangeGas exchangeHuman cells are primarily reliant on aerobic metabolism. The respiratory system is involved in pulmonary ventilation and external respiration, while the circulatory system is responsible for transport and internal respiration. Pulmonary ventilation (breathing) represents movement of air into and out of the lungs. External respiration, or gas exchange, is represented by the O2 and CO2 exchange between the lungs and the blood.Gas Exchange → chronic hypoxic respiratory failureRespiratory failureRespiratory failure is a syndrome that develops when the respiratory system is unable to maintain oxygenation and/or ventilation. Respiratory failure may be acute or chronic and is classified as hypoxemic, hypercapnic, or a combination of the two. Respiratory Failure
Clinical Presentation
Symptoms
PatientsPatientsIndividuals participating in the health care system for the purpose of receiving therapeutic, diagnostic, or preventive procedures.Clinician–Patient Relationship with IPF can have variableVariableVariables represent information about something that can change. The design of the measurement scales, or of the methods for obtaining information, will determine the data gathered and the characteristics of that data. As a result, a variable can be qualitative or quantitative, and may be further classified into subgroups.Types of Variables presentations, and early in its course, IPF can be asymptomatic.
Presentation of symptoms suggests a moderate to advanced stage:
Associated systemic symptoms are uncommon but can include:
FatigueFatigueThe state of weariness following a period of exertion, mental or physical, characterized by a decreased capacity for work and reduced efficiency to respond to stimuli.Fibromyalgia
Bibasilar fine inspiratory (“Velcro-like”) crackles on auscultation
End-inspiratory “squeaks” in advanced disease with bronchiectasisBronchiectasisBronchiectasis is a chronic disease of the airways that results from permanent bronchial distortion. This results from a continuous cycle of inflammation, bronchial damage and dilation, impaired clearance of secretions, and recurrent infections. Bronchiectasis
PatientsPatientsIndividuals participating in the health care system for the purpose of receiving therapeutic, diagnostic, or preventive procedures.Clinician–Patient Relationship can present with pulmonary hypertensionPulmonary HypertensionPulmonary hypertension (PH) or pulmonary arterial hypertension (PAH) is characterized by elevated pulmonary arterial pressure, which can lead to chronic progressive right heart failure. Pulmonary hypertension is grouped into 5 categories based on etiology, which include primary PAH, and PH due to cardiac disease, lung or hypoxic disease, chronic thromboembolic disease, and multifactorial or unclear etiologies. Pulmonary Hypertension (PHpHThe quantitative measurement of the acidity or basicity of a solution.Acid-Base Balance) and cor pulmonaleCor PulmonaleCor pulmonale is right ventricular (RV) dysfunction caused by lung disease that results in pulmonary artery hypertension. The most common cause of cor pulmonale is chronic obstructive pulmonary disease. Dyspnea is the usual presenting symptom. Cor Pulmonale:
Pitting edemaPitting edemaEdema caused by excess fluid without excess colloid. Leaves “pits” due to fluid displacement when pressure is applied to the areaEdema (pedal or sacral)
Jugular venous distention
CyanosisCyanosisA bluish or purplish discoloration of the skin and mucous membranes due to an increase in the amount of deoxygenated hemoglobin in the blood or a structural defect in the hemoglobin molecule.Pulmonary Examination
Split second heart sound with dominant P2 component
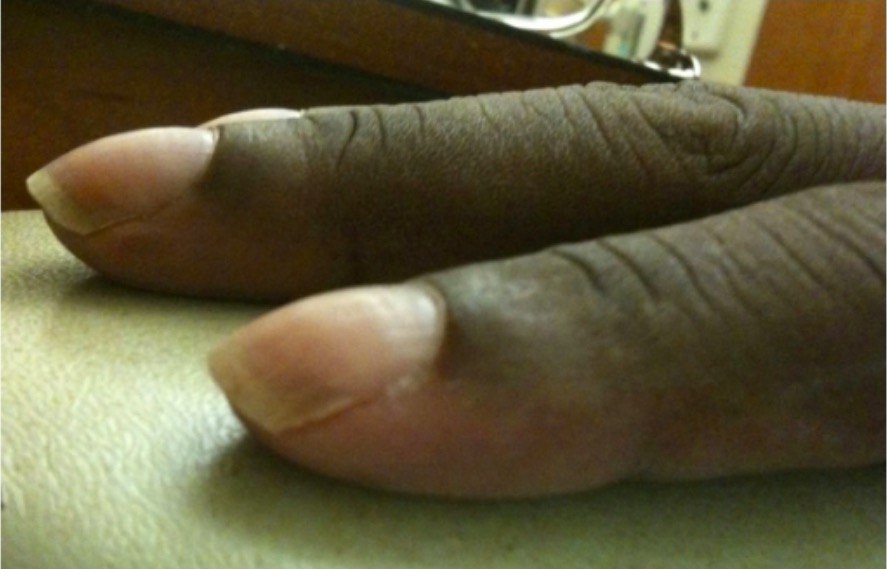
Nail clubbing:
Clubbed nails are abnormal, rounded nail beds often seen in association with conditions causing chronic hypoxemia, such as cystic fibrosis or interstitial lung disease.
Image: “Nail clubbing” by Department of Internal Medicine, Cleveland Clinic Florida, 2950 Cleveland Clinic Boulevard, Weston, FL 33331, USA. License: CC BY 3.0
IdiopathicIdiopathicDermatomyositis pulmonary fibrosisFibrosisAny pathological condition where fibrous connective tissue invades any organ, usually as a consequence of inflammation or other injury.Bronchiolitis Obliterans is diagnosed by a combination of radiologic, pathologic, and clinical investigations.
Important patient history
It is critical to obtain the following on top of a complete medical history to ensure that other causes of ILDILDInterstitial lung diseases are a heterogeneous group of disorders characterized by the inflammation and fibrosis of lung parenchyma, especially the pulmonary connective tissue in the alveolar walls. It may be idiopathic (e.g., idiopathic pulmonary fibrosis) or secondary to connective tissue diseases, medications, malignancies, occupational exposure, or allergens.Interstitial Lung Diseases are excluded:
RadiationRadiationEmission or propagation of acoustic waves (sound), electromagnetic energy waves (such as light; radio waves; gamma rays; or x-rays), or a stream of subatomic particles (such as electrons; neutrons; protons; or alpha particles).Osteosarcoma exposure
Notable medications associated with pulmonary fibrosisFibrosisAny pathological condition where fibrous connective tissue invades any organ, usually as a consequence of inflammation or other injury.Bronchiolitis Obliterans:
Amiodarone
BleomycinBleomycinA complex of related glycopeptide antibiotics from streptomyces verticillus consisting of bleomycin a2 and b2. It inhibits DNA metabolism and is used as an antineoplastic, especially for solid tumors.Antitumor Antibiotics
Nitrofurantoin
MethotrexateMethotrexateAn antineoplastic antimetabolite with immunosuppressant properties. It is an inhibitor of tetrahydrofolate dehydrogenase and prevents the formation of tetrahydrofolate, necessary for synthesis of thymidylate, an essential component of DNA.Antimetabolite Chemotherapy
Pulmonary function tests
Typical findings:
Restrictive ventilatory defect
↓ Diffusing capacity of the lung for carbon monoxideCarbon monoxideCarbon monoxide (CO). A poisonous colorless, odorless, tasteless gas. It combines with hemoglobin to form carboxyhemoglobin, which has no oxygen carrying capacity. The resultant oxygen deprivation causes headache, dizziness, decreased pulse and respiratory rates, unconsciousness, and death.Carbon Monoxide Poisoning (DLCODLCOPulmonary Function Tests)
Measurements:
Static lung volumes using body plethysmography typically reveal ↓ lung volumes (restriction) such as:
Functional residual capacityFunctional residual capacityThe volume of air remaining in the lungs at the end of a normal, quiet expiration. It is the sum of the residual volume and the expiratory reserve volume. Common abbreviation is frc.Ventilation: Mechanics of Breathing (FRC)
Total lung capacityTotal lung capacityThe volume of air contained in the lungs at the end of a maximal inspiration. It is the equivalent to each of the following sums: vital capacity plus residual volume; inspiratory capacity plus functional residual capacity; tidal volume plus inspiratory reserve volume plus functional residual capacity; or tidal volume plus inspiratory reserve volume plus expiratory reserve volume plus residual volume.Ventilation: Mechanics of Breathing (TLC)
Normal or ↑ forced expiratory volume in 1 second (FEV1)/FVC ratio
Static pressure–volume curve shifted downward and to the right → ↓ lung complianceComplianceDistensibility measure of a chamber such as the lungs (lung compliance) or bladder. Compliance is expressed as a change in volume per unit change in pressure.Veins: Histology
Imaging
Chest X-rayX-rayPenetrating electromagnetic radiation emitted when the inner orbital electrons of an atom are excited and release radiant energy. X-ray wavelengths range from 1 pm to 10 nm. Hard x-rays are the higher energy, shorter wavelength x-rays. Soft x-rays or grenz rays are less energetic and longer in wavelength. The short wavelength end of the x-ray spectrum overlaps the gamma rays wavelength range. The distinction between gamma rays and x-rays is based on their radiation source.Pulmonary Function Tests:
Significantly more sensitive and specific for the diagnosis of IPF
Features (predominantly seen in the lower lobes):
Peripheral and subpleural reticular septal thickening
Traction bronchiectasisBronchiectasisBronchiectasis is a chronic disease of the airways that results from permanent bronchial distortion. This results from a continuous cycle of inflammation, bronchial damage and dilation, impaired clearance of secretions, and recurrent infections. Bronchiectasis
Honeycombing cystsCystsAny fluid-filled closed cavity or sac that is lined by an epithelium. Cysts can be of normal, abnormal, non-neoplastic, or neoplastic tissues.Fibrocystic Change
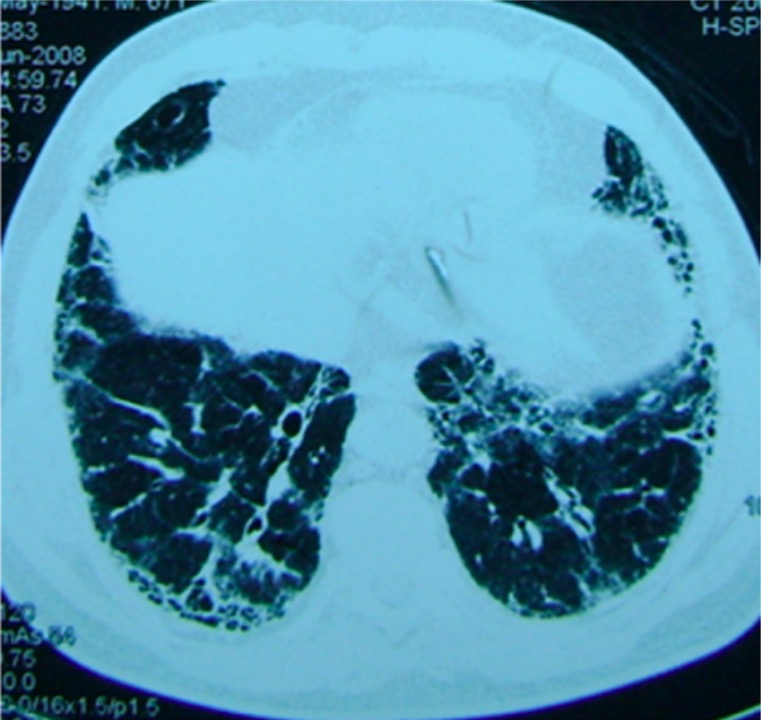
CT image from a patient with pulmonary fibrosis: A basilar reticular pattern is seen.
Image: “Patient with pulmonary fibrosis” by Department of Respiratory Medicine, Peking Union Medical College Hospital, Chinese Academy of Medical Sciences & Peking Union Medical College, #1 Shuaifuyuan Street, Beijing, Dongcheng District 100730, China. License: CC BY 2.0
Surgical specimens can be obtained using video-assisted thoracoscopic surgery (VATS). PatientsPatientsIndividuals participating in the health care system for the purpose of receiving therapeutic, diagnostic, or preventive procedures.Clinician–Patient Relationship with IPF may show a pattern of usual interstitial pneumoniaPneumoniaPneumonia or pulmonary inflammation is an acute or chronic inflammation of lung tissue. Causes include infection with bacteria, viruses, or fungi. In more rare cases, pneumonia can also be caused through toxic triggers through inhalation of toxic substances, immunological processes, or in the course of radiotherapy.Pneumonia:
Alternating zones of normal and abnormal lung tissue
FibrosisFibrosisAny pathological condition where fibrous connective tissue invades any organ, usually as a consequence of inflammation or other injury.Bronchiolitis Obliterans
Fibroblast foci (areas of active fibroproliferation)
Honeycombing patterns
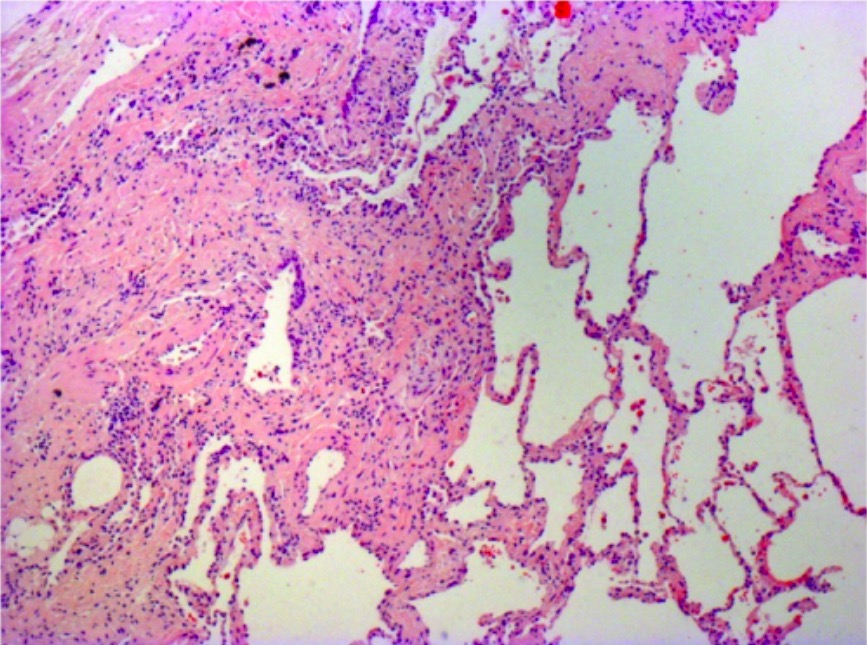
Photomicrograph of biopsy from a patient with idiopathic pulmonary fibrosis:
The typical histopathologic features of usual interstitial pneumonia are seen, characterized by spatial heterogeneity with areas of subpleural and paraseptal fibrosis and honeycombing changes (cystic airspaces lined by bronchiolar epithelium) alternating with areas of relatively spared lung parenchyma, temporal heterogeneity with admixed areas of active fibrosis with fibroblast foci, extracellular matrix deposition (mainly collagen), and relative mild or absent inflammatory cell infiltrate together with regions of histologically normal lung tissue.
Image: “Photomicrograph of biopsy from a 63-year-old man with a multi-disciplinary diagnosis of idiopathic pulmonary fibrosis” by Interstitial Lung Disease Unit, Royal Brompton and Harefield NHS Foundation Trust, London, UK. License: CC BY 4.0
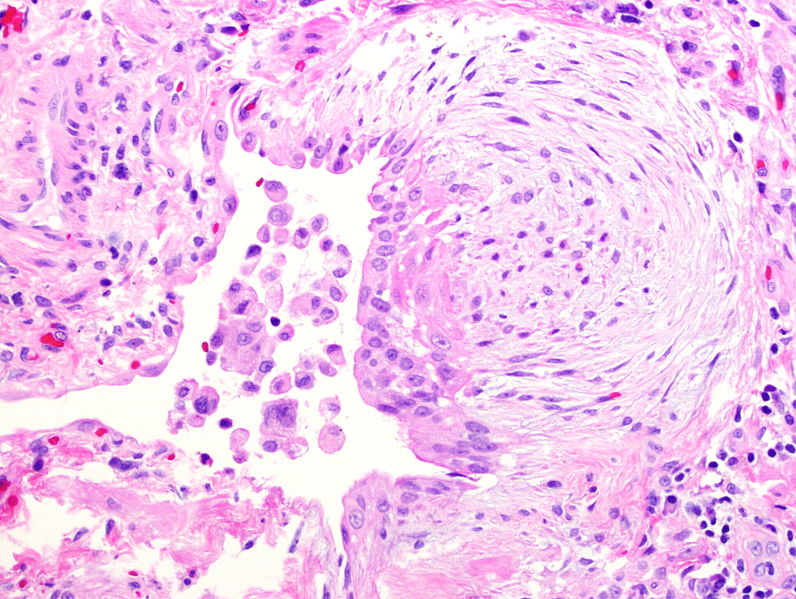
Fibroblast focus seen in idiopathic pulmonary fibrosis due to migration and proliferation of fibroblasts and myofibroblasts
Exclude alternative diagnoses if imaging is indeterminate.
Evaluate for coexisting infection.
Supporting laboratory evaluation
The following can be performed to exclude other causes of ILDILDInterstitial lung diseases are a heterogeneous group of disorders characterized by the inflammation and fibrosis of lung parenchyma, especially the pulmonary connective tissue in the alveolar walls. It may be idiopathic (e.g., idiopathic pulmonary fibrosis) or secondary to connective tissue diseases, medications, malignancies, occupational exposure, or allergens.Interstitial Lung Diseases and will be guided by clinical suspicion:
Antinuclear antibodiesAntibodiesImmunoglobulins (Igs), also known as antibodies, are glycoprotein molecules produced by plasma cells that act in immune responses by recognizing and binding particular antigens. The various Ig classes are IgG (the most abundant), IgM, IgE, IgD, and IgA, which differ in their biologic features, structure, target specificity, and distribution.Immunoglobulins: Types and Functions
Rheumatoid factorRheumatoid factorAntibodies found in adult rheumatoid arthritis patients that are directed against gamma-chain immunoglobulins.Autoimmune Hepatitis and anti–cyclic citrullinated peptide antibodiesAntibodiesImmunoglobulins (Igs), also known as antibodies, are glycoprotein molecules produced by plasma cells that act in immune responses by recognizing and binding particular antigens. The various Ig classes are IgG (the most abundant), IgM, IgE, IgD, and IgA, which differ in their biologic features, structure, target specificity, and distribution.Immunoglobulins: Types and Functions
Myositis-specific antibodiesAntibodiesImmunoglobulins (Igs), also known as antibodies, are glycoprotein molecules produced by plasma cells that act in immune responses by recognizing and binding particular antigens. The various Ig classes are IgG (the most abundant), IgM, IgE, IgD, and IgA, which differ in their biologic features, structure, target specificity, and distribution.Immunoglobulins: Types and Functions
Anti-SSA/Ro and anti-SSB/La antibodiesAntibodiesImmunoglobulins (Igs), also known as antibodies, are glycoprotein molecules produced by plasma cells that act in immune responses by recognizing and binding particular antigens. The various Ig classes are IgG (the most abundant), IgM, IgE, IgD, and IgA, which differ in their biologic features, structure, target specificity, and distribution.Immunoglobulins: Types and Functions
There are limited options for pharmacologic treatment in patientsPatientsIndividuals participating in the health care system for the purpose of receiving therapeutic, diagnostic, or preventive procedures.Clinician–Patient Relationship with IPF, and none provide a cure.
Diuresis is used to maintain euvolemiaEuvolemiaVolume Depletion and Dehydration in the presence of right heart failureHeart FailureA heterogeneous condition in which the heart is unable to pump out sufficient blood to meet the metabolic need of the body. Heart failure can be caused by structural defects, functional abnormalities (ventricular dysfunction), or a sudden overload beyond its capacity. Chronic heart failure is more common than acute heart failure which results from sudden insult to cardiac function, such as myocardial infarction.Total Anomalous Pulmonary Venous Return (TAPVR).
GERDGERDGastroesophageal reflux disease (GERD) occurs when the stomach acid frequently flows back into the esophagus. This backwash (acid reflux) can irritate the lining of the esophagus, causing symptoms such as retrosternal burning pain (heartburn). Gastroesophageal Reflux Disease (GERD) management:
Long-term oxygen therapy when patientsPatientsIndividuals participating in the health care system for the purpose of receiving therapeutic, diagnostic, or preventive procedures.Clinician–Patient Relationship resting or ambulatory saturations are < 88%
Pulmonary rehabilitation:
Alleviate the overt symptoms.
Improve functional status.
May include:
Exercise training
SmokingSmokingWillful or deliberate act of inhaling and exhaling smoke from burning substances or agents held by hand.Interstitial Lung Diseases cessation
Psychosocial assistance
Supportive care
Vaccinations:
InfluenzaInfluenzaInfluenza viruses are members of the Orthomyxoviridae family and the causative organisms of influenza, a highly contagious febrile respiratory disease. There are 3 primary influenza viruses (A, B, and C) and various subtypes, which are classified based on their virulent surface antigens, hemagglutinin (HA) and neuraminidase (NA). Influenza typically presents with a fever, myalgia, headache, and symptoms of an upper respiratory infection. Influenza Viruses/Influenza
PneumoniaPneumoniaPneumonia or pulmonary inflammation is an acute or chronic inflammation of lung tissue. Causes include infection with bacteria, viruses, or fungi. In more rare cases, pneumonia can also be caused through toxic triggers through inhalation of toxic substances, immunological processes, or in the course of radiotherapy.Pneumonia
Bilateral lung transplantationLung transplantationThe transference of either one or both of the lungs from one human or animal to another.Organ Transplantation is more commonly used than single lung transplantationLung transplantationThe transference of either one or both of the lungs from one human or animal to another.Organ Transplantation.
Respiratory infectionsInfectionsInvasion of the host organism by microorganisms or their toxins or by parasites that can cause pathological conditions or diseases.Chronic Granulomatous Disease
Thromboembolic disease
Lung cancerLung cancerLung cancer is the malignant transformation of lung tissue and the leading cause of cancer-related deaths. The majority of cases are associated with long-term smoking. The disease is generally classified histologically as either small cell lung cancer or non-small cell lung cancer. Symptoms include cough, dyspnea, weight loss, and chest discomfort. Lung Cancer
Type 3Type 3Spinal Muscular AtrophyPHpHThe quantitative measurement of the acidity or basicity of a solution.Acid-Base Balance and cor pulmonaleCor PulmonaleCor pulmonale is right ventricular (RV) dysfunction caused by lung disease that results in pulmonary artery hypertension. The most common cause of cor pulmonale is chronic obstructive pulmonary disease. Dyspnea is the usual presenting symptom. Cor Pulmonale
Respiratory failureRespiratory failureRespiratory failure is a syndrome that develops when the respiratory system is unable to maintain oxygenation and/or ventilation. Respiratory failure may be acute or chronic and is classified as hypoxemic, hypercapnic, or a combination of the two. Respiratory Failure
PrognosisPrognosisA prediction of the probable outcome of a disease based on a individual’s condition and the usual course of the disease as seen in similar situations.Non-Hodgkin Lymphomas
IPF progression is associated with an estimated median survival time of up to 5 years following the diagnosis.
The rate of decline and progression to death in patientsPatientsIndividuals participating in the health care system for the purpose of receiving therapeutic, diagnostic, or preventive procedures.Clinician–Patient Relationship with IPF may take several clinical forms:
Slow physiologic deterioration with worsening severity of dyspneaDyspneaDyspnea is the subjective sensation of breathing discomfort. Dyspnea is a normal manifestation of heavy physical or psychological exertion, but also may be caused by underlying conditions (both pulmonary and extrapulmonary). Dyspnea
Rapid deterioration and progression to death
Periods of relative stability interposed with periods of acute respiratory decline
Acute respiratory failureRespiratory failureRespiratory failure is a syndrome that develops when the respiratory system is unable to maintain oxygenation and/or ventilation. Respiratory failure may be acute or chronic and is classified as hypoxemic, hypercapnic, or a combination of the two. Respiratory Failure is a common cause of hospital admission and death, and can result from:
Disease progression
PneumoniaPneumoniaPneumonia or pulmonary inflammation is an acute or chronic inflammation of lung tissue. Causes include infection with bacteria, viruses, or fungi. In more rare cases, pneumonia can also be caused through toxic triggers through inhalation of toxic substances, immunological processes, or in the course of radiotherapy.Pneumonia
Pulmonary hypertensionPulmonary HypertensionPulmonary hypertension (PH) or pulmonary arterial hypertension (PAH) is characterized by elevated pulmonary arterial pressure, which can lead to chronic progressive right heart failure. Pulmonary hypertension is grouped into 5 categories based on etiology, which include primary PAH, and PH due to cardiac disease, lung or hypoxic disease, chronic thromboembolic disease, and multifactorial or unclear etiologies. Pulmonary Hypertension
Differential Diagnosis
SarcoidosisSarcoidosisSarcoidosis is a multisystem inflammatory disease that causes noncaseating granulomas. The exact etiology is unknown. Sarcoidosis usually affects the lungs and thoracic lymph nodes, but it can also affect almost every system in the body, including the skin, heart, and eyes, most commonly. Sarcoidosis: granulomatous disorder affecting multiple organ systems without a known etiology: The most common presenting findings include pulmonary reticular opacities, bilateral hilar adenopathy, and skinSkinThe skin, also referred to as the integumentary system, is the largest organ of the body. The skin is primarily composed of the epidermis (outer layer) and dermis (deep layer). The epidermis is primarily composed of keratinocytes that undergo rapid turnover, while the dermis contains dense layers of connective tissue.Skin: Structure and Functions, joint, or eye lesions. PatientsPatientsIndividuals participating in the health care system for the purpose of receiving therapeutic, diagnostic, or preventive procedures.Clinician–Patient Relationship are often asymptomatic, though may present with cough, dyspneaDyspneaDyspnea is the subjective sensation of breathing discomfort. Dyspnea is a normal manifestation of heavy physical or psychological exertion, but also may be caused by underlying conditions (both pulmonary and extrapulmonary). Dyspnea, feverFeverFever is defined as a measured body temperature of at least 38°C (100.4°F). Fever is caused by circulating endogenous and/or exogenous pyrogens that increase levels of prostaglandin E2 in the hypothalamus. Fever is commonly associated with chills, rigors, sweating, and flushing of the skin. Fever, and malaiseMalaiseTick-borne Encephalitis Virus. Diagnosis involves imaging, elevated serum ACE levels, and BAL and often requires a biopsyBiopsyRemoval and pathologic examination of specimens from the living body.Ewing Sarcoma. Management is usually with glucocorticoidsGlucocorticoidsGlucocorticoids are a class within the corticosteroid family. Glucocorticoids are chemically and functionally similar to endogenous cortisol. There are a wide array of indications, which primarily benefit from the antiinflammatory and immunosuppressive effects of this class of drugs.Glucocorticoids.
Hypersensitivity pneumonitisPneumonitisHuman Herpesvirus 6 and 7: immunologically induced inflammatory disease affecting the alveoliAlveoliSmall polyhedral outpouchings along the walls of the alveolar sacs, alveolar ducts and terminal bronchioles through the walls of which gas exchange between alveolar air and pulmonary capillary blood takes place.Acute Respiratory Distress Syndrome (ARDS), bronchiolesBronchiolesThe small airways branching off the tertiary bronchi. Terminal bronchioles lead into several orders of respiratory bronchioles which in turn lead into alveolar ducts and then into pulmonary alveoli.Bronchial Tree: Anatomy, and lung parenchyma resulting from exposure to inhaled antigens: PatientsPatientsIndividuals participating in the health care system for the purpose of receiving therapeutic, diagnostic, or preventive procedures.Clinician–Patient Relationship may develop cough, dyspneaDyspneaDyspnea is the subjective sensation of breathing discomfort. Dyspnea is a normal manifestation of heavy physical or psychological exertion, but also may be caused by underlying conditions (both pulmonary and extrapulmonary). Dyspnea, and fatigueFatigueThe state of weariness following a period of exertion, mental or physical, characterized by a decreased capacity for work and reduced efficiency to respond to stimuli.Fibromyalgia. HRCTHRCTPulmonary Function Tests will show diffuse, poorly defined centrilobular micronodules or ground-glass opacities. Pulmonary function tests are variableVariableVariables represent information about something that can change. The design of the measurement scales, or of the methods for obtaining information, will determine the data gathered and the characteristics of that data. As a result, a variable can be qualitative or quantitative, and may be further classified into subgroups.Types of Variables. Management includes avoiding the inciting agent and administering steroidsSteroidsA group of polycyclic compounds closely related biochemically to terpenes. They include cholesterol, numerous hormones, precursors of certain vitamins, bile acids, alcohols (sterols), and certain natural drugs and poisons. Steroids have a common nucleus, a fused, reduced 17-carbon atom ring system, cyclopentanoperhydrophenanthrene. Most steroids also have two methyl groups and an aliphatic side-chain attached to the nucleus.Benign Liver Tumors in subacute and chronic cases.
PneumoconiosisPneumoconiosisPneumoconiosis is an occupational disease that results from the inhalation and deposition of mineral dusts and other inorganic particles in the lung. It can be categorized according to the type of causative particle involved or by the type of response provoked.Pneumoconiosis: occupational disease that results from inhalation of inorganic particles into the lungsLungsLungs are the main organs of the respiratory system. Lungs are paired viscera located in the thoracic cavity and are composed of spongy tissue. The primary function of the lungs is to oxygenate blood and eliminate CO2. Lungs: Anatomy: In the lungsLungsLungs are the main organs of the respiratory system. Lungs are paired viscera located in the thoracic cavity and are composed of spongy tissue. The primary function of the lungs is to oxygenate blood and eliminate CO2. Lungs: Anatomy, these particles can cause chronic inflammationChronic InflammationInflammation and fibrosisFibrosisAny pathological condition where fibrous connective tissue invades any organ, usually as a consequence of inflammation or other injury.Bronchiolitis Obliterans. PatientsPatientsIndividuals participating in the health care system for the purpose of receiving therapeutic, diagnostic, or preventive procedures.Clinician–Patient Relationship will have progressive dyspneaDyspneaDyspnea is the subjective sensation of breathing discomfort. Dyspnea is a normal manifestation of heavy physical or psychological exertion, but also may be caused by underlying conditions (both pulmonary and extrapulmonary). Dyspnea and dry coughDry CoughStrongyloidiasis. Chest X-rayX-rayPenetrating electromagnetic radiation emitted when the inner orbital electrons of an atom are excited and release radiant energy. X-ray wavelengths range from 1 pm to 10 nm. Hard x-rays are the higher energy, shorter wavelength x-rays. Soft x-rays or grenz rays are less energetic and longer in wavelength. The short wavelength end of the x-ray spectrum overlaps the gamma rays wavelength range. The distinction between gamma rays and x-rays is based on their radiation source.Pulmonary Function Tests findings can vary depending on the causative particle, but may include ground-glass opacities, calcifications, lung nodules, and pleural irregularities. Management is mainly symptomatic.
Chronic obstructive pulmonary diseasePulmonary diseaseDiseases involving the respiratory system.Blastomyces/Blastomycosis (COPDCOPDChronic obstructive pulmonary disease (COPD) is a lung disease characterized by progressive, largely irreversible airflow obstruction. The condition usually presents in middle-aged or elderly persons with a history of cigarette smoking. Signs and symptoms include prolonged expiration, wheezing, diminished breath sounds, progressive dyspnea, and chronic cough. Chronic Obstructive Pulmonary Disease (COPD)): lung disease characterized by progressive, largely irreversible airflow obstruction: Symptoms include progressive dyspneaDyspneaDyspnea is the subjective sensation of breathing discomfort. Dyspnea is a normal manifestation of heavy physical or psychological exertion, but also may be caused by underlying conditions (both pulmonary and extrapulmonary). Dyspnea and chronic cough. Prolonged expirationExpirationVentilation: Mechanics of Breathing, wheezingWheezingWheezing is an abnormal breath sound characterized by a whistling noise that can be relatively high-pitched and shrill (more common) or coarse. Wheezing is produced by the movement of air through narrowed or compressed small (intrathoracic) airways. Wheezing, and/or diminished breath sounds may be noted on physical exam. Unlike IPF, pulmonary function testingPulmonary Function TestingPulmonary Function Tests is consistent with obstruction. Management includes smokingSmokingWillful or deliberate act of inhaling and exhaling smoke from burning substances or agents held by hand.Interstitial Lung Diseases cessation, pulmonary rehabilitation, and pharmacotherapy.
Heart failureHeart FailureA heterogeneous condition in which the heart is unable to pump out sufficient blood to meet the metabolic need of the body. Heart failure can be caused by structural defects, functional abnormalities (ventricular dysfunction), or a sudden overload beyond its capacity. Chronic heart failure is more common than acute heart failure which results from sudden insult to cardiac function, such as myocardial infarction.Total Anomalous Pulmonary Venous Return (TAPVR): inability to produce normal cardiac outputCardiac outputThe volume of blood passing through the heart per unit of time. It is usually expressed as liters (volume) per minute so as not to be confused with stroke volume (volume per beat).Cardiac Mechanics to meet metabolic needs: PatientsPatientsIndividuals participating in the health care system for the purpose of receiving therapeutic, diagnostic, or preventive procedures.Clinician–Patient Relationship present with dyspneaDyspneaDyspnea is the subjective sensation of breathing discomfort. Dyspnea is a normal manifestation of heavy physical or psychological exertion, but also may be caused by underlying conditions (both pulmonary and extrapulmonary). Dyspnea, hypoxiaHypoxiaSub-optimal oxygen levels in the ambient air of living organisms.Ischemic Cell Damage, and peripheral edemaPeripheral edemaPeripheral edema is the swelling of the lower extremities, namely, legs, feet, and ankles.Edema. BNPBNPA peptide that is secreted by the brain and the heart atria, stored mainly in cardiac ventricular myocardium. It can cause natriuresis; diuresis; vasodilation; and inhibits secretion of renin and aldosterone. It improves heart function. It contains 32 amino acids.Renal Sodium and Water Regulation will be elevated, and pulmonary edemaPulmonary edemaPulmonary edema is a condition caused by excess fluid within the lung parenchyma and alveoli as a consequence of a disease process. Based on etiology, pulmonary edema is classified as cardiogenic or noncardiogenic. Patients may present with progressive dyspnea, orthopnea, cough, or respiratory failure.Pulmonary Edema may be seen on X-rayX-rayPenetrating electromagnetic radiation emitted when the inner orbital electrons of an atom are excited and release radiant energy. X-ray wavelengths range from 1 pm to 10 nm. Hard x-rays are the higher energy, shorter wavelength x-rays. Soft x-rays or grenz rays are less energetic and longer in wavelength. The short wavelength end of the x-ray spectrum overlaps the gamma rays wavelength range. The distinction between gamma rays and x-rays is based on their radiation source.Pulmonary Function Tests examination. EchocardiographyEchocardiographyUltrasonic recording of the size, motion, and composition of the heart and surrounding tissues. The standard approach is transthoracic.Tricuspid Valve Atresia (TVA) will confirm the diagnosis. Management relies on diuresis and medical optimization of cardiac function with beta-blockersBeta-blockersDrugs that bind to but do not activate beta-adrenergic receptors thereby blocking the actions of beta-adrenergic agonists. Adrenergic beta-antagonists are used for treatment of hypertension, cardiac arrhythmias, angina pectoris, glaucoma, migraine headaches, and anxiety.Class 2 Antiarrhythmic Drugs (Beta Blockers) and ACE inhibitorsACE inhibitorsTruncus Arteriosus.
Lederer, D. J., Martinez, F. J. (2018). Idiopathic pulmonary fibrosis. New England Journal of Medicine 378:1811–1823.
Waxman, A., Restrepo-Jaramillo, R., Thenappan, T., Ravichandran, A., Engel, P., Bajwa, A., Nathan, S. D. (2021). Inhaled treprostinil in pulmonary hypertension due to interstitial lung disease. New England Journal of Medicine 384:325–334.