Granulomatosis with polyangiitis (GPA, formerly known as Wegener's granulomatosis) is a rare autoimmune disease of unknown etiology. It leads to a necrotizing granulomatous inflammation Granulomatous Inflammation Chalazion of small and medium-sized blood vessels of the nose Nose The nose is the human body's primary organ of smell and functions as part of the upper respiratory system. The nose may be best known for inhaling oxygen and exhaling carbon dioxide, but it also contributes to other important functions, such as tasting. The anatomy of the nose can be divided into the external nose and the nasal cavity. Nose Anatomy (External & Internal), sinuses, throat Throat The pharynx is a component of the digestive system that lies posterior to the nasal cavity, oral cavity, and larynx. The pharynx can be divided into the oropharynx, nasopharynx, and laryngopharynx. Pharyngeal muscles play an integral role in vital processes such as breathing, swallowing, and speaking. Pharynx: Anatomy, lungs Lungs Lungs are the main organs of the respiratory system. Lungs are paired viscera located in the thoracic cavity and are composed of spongy tissue. The primary function of the lungs is to oxygenate blood and eliminate CO2. Lungs: Anatomy, and kidneys Kidneys The kidneys are a pair of bean-shaped organs located retroperitoneally against the posterior wall of the abdomen on either side of the spine. As part of the urinary tract, the kidneys are responsible for blood filtration and excretion of water-soluble waste in the urine. Kidneys: Anatomy. Early stages of GPA often present with localized manifestations such as infections Infections Invasion of the host organism by microorganisms or their toxins or by parasites that can cause pathological conditions or diseases. Chronic Granulomatous Disease of the upper respiratory tract, skin Skin The skin, also referred to as the integumentary system, is the largest organ of the body. The skin is primarily composed of the epidermis (outer layer) and dermis (deep layer). The epidermis is primarily composed of keratinocytes that undergo rapid turnover, while the dermis contains dense layers of connective tissue. Skin: Structure and Functions lesions, and/or constitutional symptoms Constitutional Symptoms Antineutrophil Cytoplasmic Antibody (ANCA)-Associated Vasculitis. Later stages can present with renal failure Renal failure Conditions in which the kidneys perform below the normal level in the ability to remove wastes, concentrate urine, and maintain electrolyte balance; blood pressure; and calcium metabolism. Renal insufficiency can be classified by the degree of kidney damage (as measured by the level of proteinuria) and reduction in glomerular filtration rate. Crush Syndrome and severe respiratory disease. Early diagnosis and treatment of granulomatosis with polyangiitis (which involves the administration of corticosteroids Corticosteroids Chorioretinitis and immunosuppressive agents such as methotrexate Methotrexate An antineoplastic antimetabolite with immunosuppressant properties. It is an inhibitor of tetrahydrofolate dehydrogenase and prevents the formation of tetrahydrofolate, necessary for synthesis of thymidylate, an essential component of DNA. Antimetabolite Chemotherapy) may lead to a full remission Remission A spontaneous diminution or abatement of a disease over time, without formal treatment. Cluster Headaches but without treatment, the condition has a high mortality Mortality All deaths reported in a given population. Measures of Health Status rate.
Last updated: Dec 15, 2025
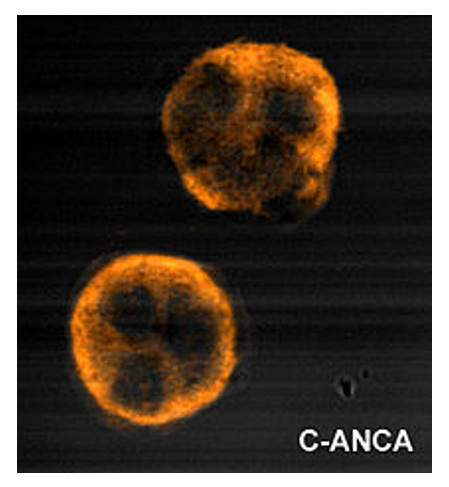
Uniform granular cytoplasmic staining of c-ANCA
Image: “C anca” by Malittle. License: CC0 1.0
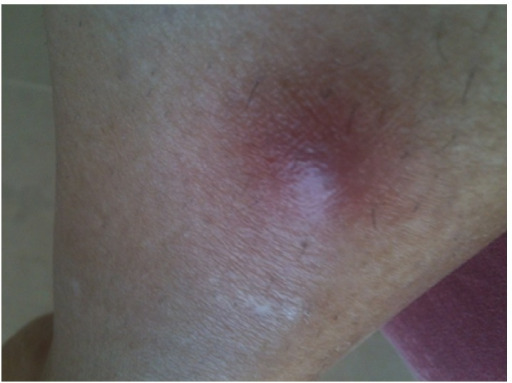
Erythematous nodule on the right shin in a patient with GPA
Image: “Initial lesion: erythematous nodule on right shin” by Section of Internal Medicine, Department of Medicine, The Aga Khan University Hospital, P.O. Box 3500, Stadium Road, Karachi 74800, Pakistan. License: CC BY 3.0
Example of a collapsed nasal bridge (saddle nose deformity) seen in GPA. The patient also shows orbital involvement with proptosis (protrusion of the eyeball), eyelid edema, and limitation of ocular movements. Orbital involvement typically does not occur until years after disease onset.
Image: “Anterior and lateral appearance of the patient” by Ophthalmic Research Center, Shahid Beheshti University of Medical Sciences, Tehran, Iran. License: CC BY 2.5| 1. Nasal or oral inflammation Inflammation Inflammation is a complex set of responses to infection and injury involving leukocytes as the principal cellular mediators in the body’s defense against pathogenic organisms. Inflammation is also seen as a response to tissue injury in the process of wound healing. The 5 cardinal signs of inflammation are pain, heat, redness, swelling, and loss of function. Inflammation | Painful or painless oral ulcers Oral ulcers A loss of mucous substance of the mouth showing local excavation of the surface, resulting from the sloughing of inflammatory necrotic tissue. It is the result of a variety of causes, e.g., denture irritation, aphthous stomatitis; necrotizing gingivitis, toothbrushing, and various irritants. Chédiak-Higashi Syndrome or purulent or bloody nasal discharge |
|---|---|
| 2. Abnormal chest radiograph | Pulmonary nodules, fixed pulmonary infiltrates, or pulmonary cavities |
| 3. Abnormal urinary sediment | Microscopic haematuria with or without red cell casts (glomerulonephritis) |
| 4. Granulomatous inflammation Granulomatous Inflammation Chalazion | The biopsy Biopsy Removal and pathologic examination of specimens from the living body. Ewing Sarcoma of an artery or perivascular area shows granulomatous inflammation Granulomatous Inflammation Chalazion. |
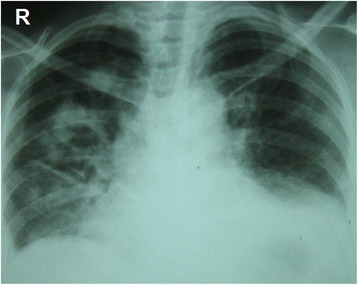
Chest radiography: Multiple nodular and cavitating lesions are visible predominately on the right side with multiple patchy infiltrates typical of granulomatosis with polyangiitis. Costophrenic angles are obliterated owing to small bi-basal pleural effusion.
Image: “Chest radiography at presentation” by Department of Pharmacology and Pharmacy, Faculty of Medicine University of Colombo, PO Box 271, Kynsey Road, Colombo 08, Sri Lanka. License: CC BY 4.0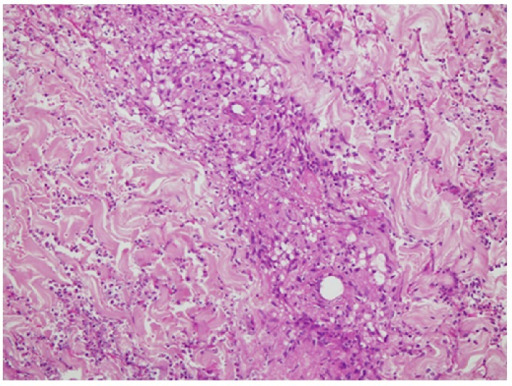
Skin biopsy: Vasculitis involving the superficial and deep dermis showing fibrinoid necrosis of vessel wall with granuloma formation, neutrophilic infiltrate, and nuclear debris.
Image: “Skin biopsy” by Section of Internal Medicine, Department of Medicine, The Aga Khan University Hospital, P.O. Box 3500, Stadium Road, Karachi 74800, Pakistan. License: CC BY 3.0