


A granulomatose com poliangeíte ( GPA GPA A multisystemic disease of a complex genetic background. It is characterized by inflammation of the blood vessels (vasculitis) leading to damage in any number of organs. The common features include granulomatous inflammation of the respiratory tract and kidneys. Most patients have measurable autoantibodies (antineutrophil cytoplasmic antibodies) against myeloblastin. Granulomatosis with Polyangiitis, anteriormente conhecida como granulomatose de Wegener) é uma doença autoimune rara de etiologia desconhecida. Caracteriza-se por uma inflamação granulomatosa necrosante dos pequenos e médios vasos sanguíneos do nariz, seios nasais, faringe, laringe, pulmões e rins. Os estadios iniciais da GPA GPA A multisystemic disease of a complex genetic background. It is characterized by inflammation of the blood vessels (vasculitis) leading to damage in any number of organs. The common features include granulomatous inflammation of the respiratory tract and kidneys. Most patients have measurable autoantibodies (antineutrophil cytoplasmic antibodies) against myeloblastin. Granulomatosis with Polyangiitis apresentam-se frequentemente com manifestações localizadas, como infeções do trato respiratório superior, lesões cutâneas e/ou sintomas constitucionais. Nas fases mais MAIS Androgen Insensitivity Syndrome tardias pode apresentar insuficiência renal e doença respiratória grave. O diagnóstico e tratamento precoce da granulomatose com poliangeíte (que envolve corticoterapia e imunossupressão com metotrexato, por exemplo) pode levar à remissão completa, no entanto, sem tratamento, a condição apresenta uma elevada taxa de mortalidade.
Last updated: Dec 15, 2025
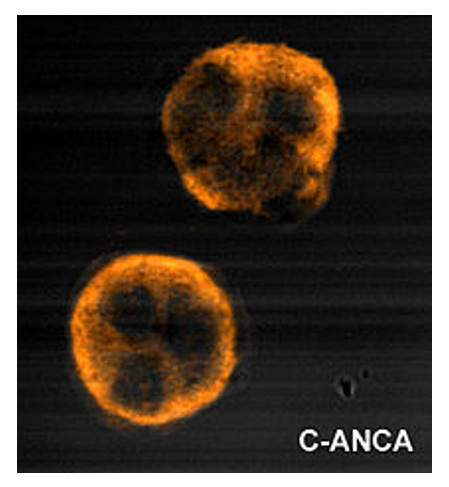
Coloração uniforme citoplasmática granular do c-ANCA
Imagem por Malittle, PD.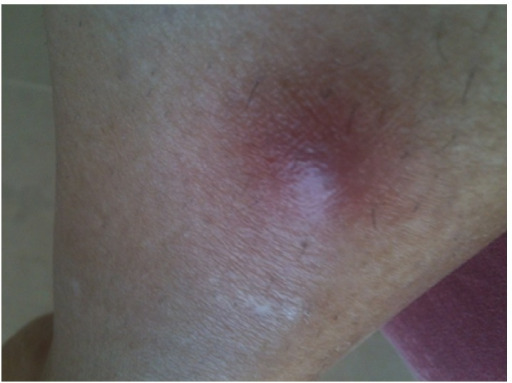
Nódulo eritematoso no membro inferior direito de um paciente com GPA
Imagem: “Initial lesion: erythematous nodule on right shin” por Section of Internal Medicine, Department of Medicine, The Aga Khan University Hospital, P.O. Box 3500, Stadium Road, Karachi 74800, Pakistan. Licença: CC BY 3.0
Exemplo de ponte nasal colapsada (deformação nasal em sela) observada na GPA. O paciente pode manifestar envolvimento ocular com proptose (protrusão do globo ocular), edema das pálpebras e limitação dos movimentos oculares. O envolvimento ocular habitualmente só ocorre anos após o início da doença.
Imagem: “Anterior and lateral appearance of the patient” por Ophthalmic Research Center, Shahid Beheshti University of Medical Sciences, Tehran, Iran. Licença: CC BY 2.5| 1. Inflamação nasal ou oral | Úlceras orais dolorosas ou indolores ou secreção nasal purulenta ou hemoptóica |
|---|---|
| 2. Radiografia do tórax anormal | Nódulos pulmonares, infiltrados pulmonares fixos, ou cavidades pulmonares |
| 3. Sedimento urinário anormal | Hematúria microscópica com ou sem cilindros eritrocitários (glomerulonefrite) |
| 4. Inflamação granulomatosa | A biópsia de uma artéria ou área perivascular revela inflamação granulomatosa. |
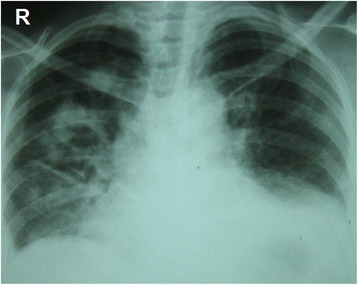
Radiografia do tórax: Múltiplas lesões nodulares e cavitárias visíveis predominantemente no lado direito, com múltiplos infiltrados irregulares típicos da granulomatose com poliangeíte. Apagamento dos ângulos costofrénicos devido a pequenos derrames pleurais bibasais.
Imagem: “Chest radiography at presentation” por Department of Pharmacology and Pharmacy, Faculty of Medicine University of Colombo, PO Box 271, Kynsey Road, Colombo 08, Sri Lanka. Licença: CC BY 4.0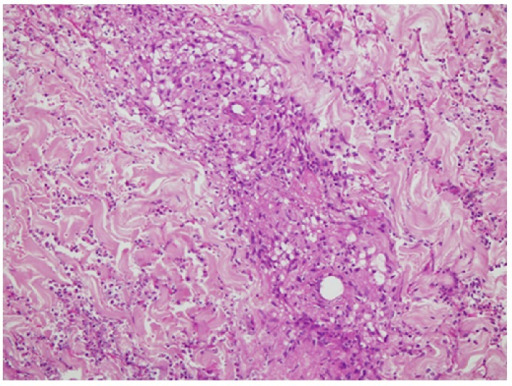
Biópsia de pele: Vasculite a envolver a derme superficial e profunda com necrose fibrinóide da parede do vaso e formação de granuloma, infiltrado neutrofílico e detritos nucleares.
Imagem: “Skin biopsy” por Section of Internal Medicine, Department of Medicine, The Aga Khan University Hospital, P.O. Box 3500, Stadium Road, Karachi 74800, Pakistan. Licença: CC BY 3.0