Mitochondrial myopathies are conditions arising from dysfunction of the mitochondria Mitochondria Semiautonomous, self-reproducing organelles that occur in the cytoplasm of all cells of most, but not all, eukaryotes. Each mitochondrion is surrounded by a double limiting membrane. The inner membrane is highly invaginated, and its projections are called cristae. Mitochondria are the sites of the reactions of oxidative phosphorylation, which result in the formation of ATP. They contain distinctive ribosomes, transfer RNAs; amino Acyl tRNA synthetases; and elongation and termination factors. Mitochondria depend upon genes within the nucleus of the cells in which they reside for many essential messenger RNAs. Mitochondria are believed to have arisen from aerobic bacteria that established a symbiotic relationship with primitive protoeukaryotes. The Cell: Organelles (the energy-producing structures) and are characterized by prominent muscular symptoms and accompanied by various symptoms from organs with high energy requirements. The organs disproportionately affected include the skeletal muscles Skeletal muscles A subtype of striated muscle, attached by tendons to the skeleton. Skeletal muscles are innervated and their movement can be consciously controlled. They are also called voluntary muscles. Muscle Tissue: Histology, brain Brain The part of central nervous system that is contained within the skull (cranium). Arising from the neural tube, the embryonic brain is comprised of three major parts including prosencephalon (the forebrain); mesencephalon (the midbrain); and rhombencephalon (the hindbrain). The developed brain consists of cerebrum; cerebellum; and other structures in the brain stem. Nervous System: Anatomy, Structure, and Classification, and heart. Mitochondrial myopathies are caused by mutations in the nuclear DNA DNA A deoxyribonucleotide polymer that is the primary genetic material of all cells. Eukaryotic and prokaryotic organisms normally contain DNA in a double-stranded state, yet several important biological processes transiently involve single-stranded regions. DNA, which consists of a polysugar-phosphate backbone possessing projections of purines (adenine and guanine) and pyrimidines (thymine and cytosine), forms a double helix that is held together by hydrogen bonds between these purines and pyrimidines (adenine to thymine and guanine to cytosine). DNA Types and Structure or mitochondrial DNA Mitochondrial DNA Double-stranded DNA of mitochondria. In eukaryotes, the mitochondrial genome is circular and codes for ribosomal rnas, transfer rnas, and about 10 proteins. DNA Types and Structure, which typically result in reduced production of energy needed by cells. Presentation can be an isolated myopathy Myopathy Dermatomyositis, encephalomyopathy, ophthalmoplegias, or a multisystem disease. Diagnosis involves in-depth medical and family history Family History Adult Health Maintenance, along with laboratory and genetic studies. On biopsy Biopsy Removal and pathologic examination of specimens from the living body. Ewing Sarcoma, there is subsarcolemmal and intermyofibrillar proliferation of mitochondria Mitochondria Semiautonomous, self-reproducing organelles that occur in the cytoplasm of all cells of most, but not all, eukaryotes. Each mitochondrion is surrounded by a double limiting membrane. The inner membrane is highly invaginated, and its projections are called cristae. Mitochondria are the sites of the reactions of oxidative phosphorylation, which result in the formation of ATP. They contain distinctive ribosomes, transfer RNAs; amino Acyl tRNA synthetases; and elongation and termination factors. Mitochondria depend upon genes within the nucleus of the cells in which they reside for many essential messenger RNAs. Mitochondria are believed to have arisen from aerobic bacteria that established a symbiotic relationship with primitive protoeukaryotes. The Cell: Organelles seen as “ragged-red fibers.” This condition indicates a compensatory response to energy failure. There is no definitive treatment. Management consists of physical therapy Physical Therapy Becker Muscular Dystrophy and a multidisciplinary approach in addressing accompanying symptoms.
Last updated: Apr 24, 2025
Mitochondrial myopathies are diseases that arise from dysfunction of the mitochondria Mitochondria Semiautonomous, self-reproducing organelles that occur in the cytoplasm of all cells of most, but not all, eukaryotes. Each mitochondrion is surrounded by a double limiting membrane. The inner membrane is highly invaginated, and its projections are called cristae. Mitochondria are the sites of the reactions of oxidative phosphorylation, which result in the formation of ATP. They contain distinctive ribosomes, transfer RNAs; amino Acyl tRNA synthetases; and elongation and termination factors. Mitochondria depend upon genes within the nucleus of the cells in which they reside for many essential messenger RNAs. Mitochondria are believed to have arisen from aerobic bacteria that established a symbiotic relationship with primitive protoeukaryotes. The Cell: Organelles (the energy-producing structures). These diseases are characterized by prominent muscular symptoms (such as muscle weakness) and accompanied by various symptoms from organs with high energy requirements.
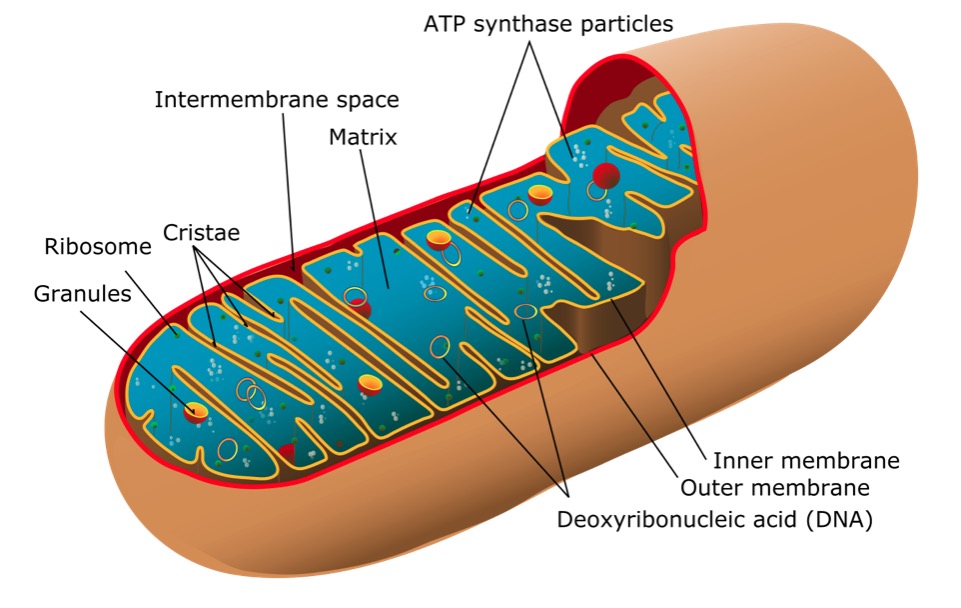
Structure of a mitochondrion
Image: “Mitochondrion of the eukaryotic cell” by Mariana Ruiz Villarreal. License: Public DomainSigns and symptoms correlate to organs or tissues affected:

Kearns-Sayre syndrome (KSS):
Patient with bilateral ptosis
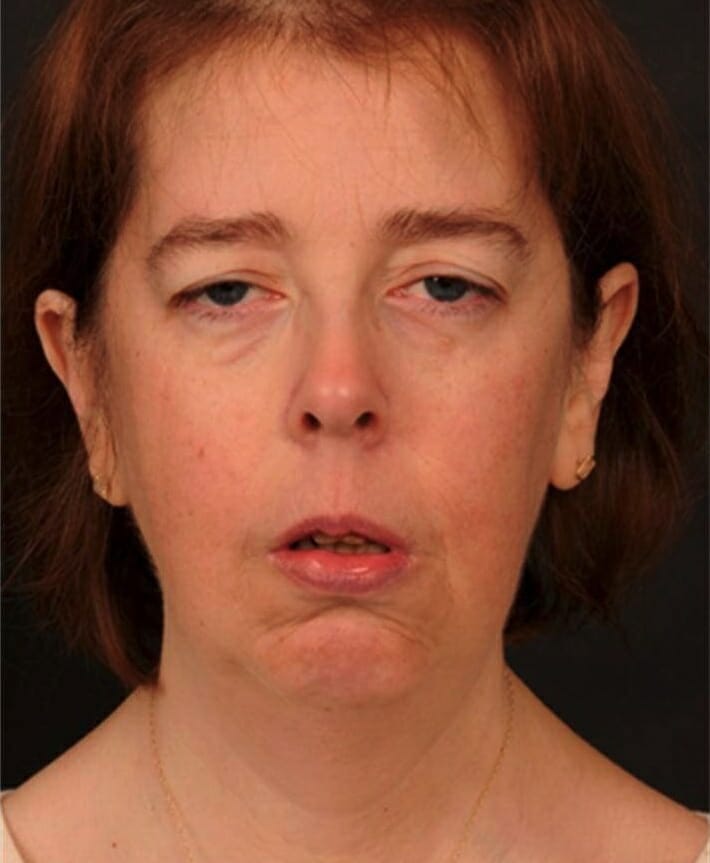
Clinical features of myoclonic epilepsy with ragged red fibers (MERRF) mutation: hair thinning, bilateral ptosis, facial diplegia with temporalis muscle wasting, jaw weakness, neck flexor muscle weakness, and neck extensor muscle weakness
Image: “Clinical features of the MERRF mutation” by Wellcome Trust Centre for Mitochondrial Research, Institute for Ageing and Health, The Medical School, Newcastle University, Newcastle upon Tyne NE2 2HH, UK. License: CC BY 3.0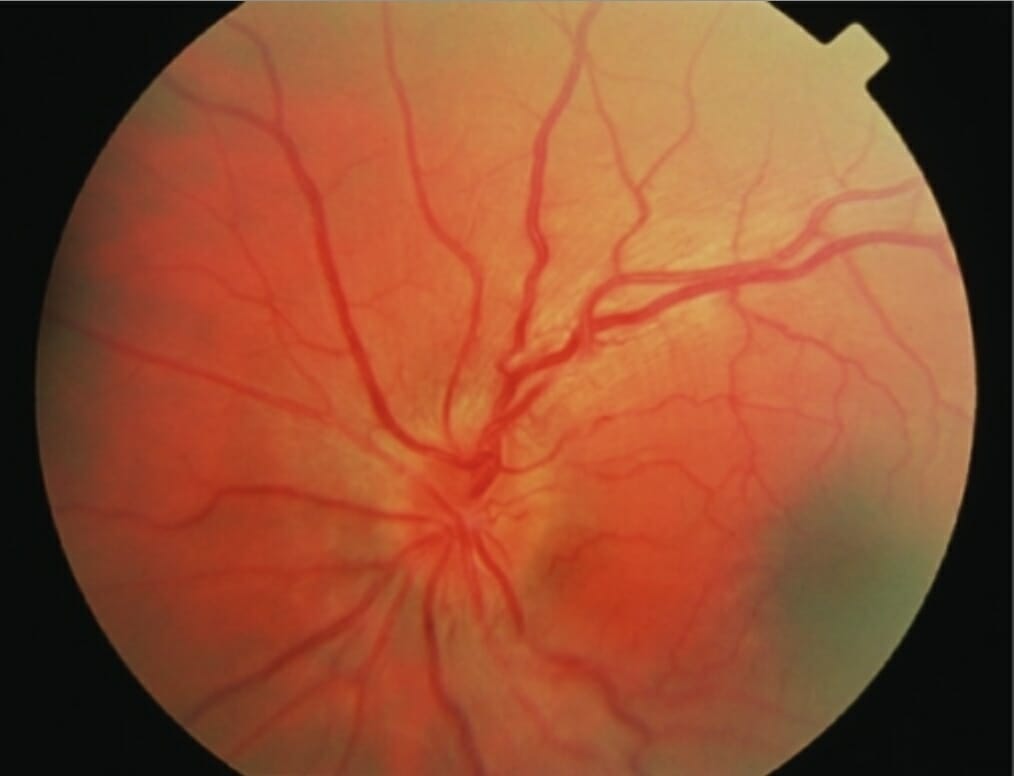
Leber hereditary optic neuropathy with disc hyperemia, retinal tortuosity, and swelling of the peripapillary retinal nerve fiber layer
Image: “Acute fundal appearance in Leber hereditary optic neuropathy” by Mitochondrial Research Group, The Medical School, Newcastle University, Newcastle upon Tyne, UK. License: CC BY 2.0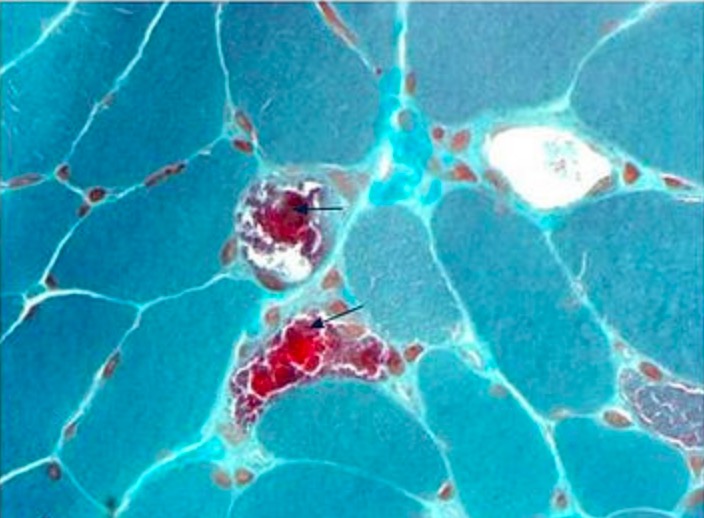
Muscle biopsy in a patient with typical features of mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes (MELAS):
Modified Gomori trichrome stain showing several ragged-red fibers (arrows)
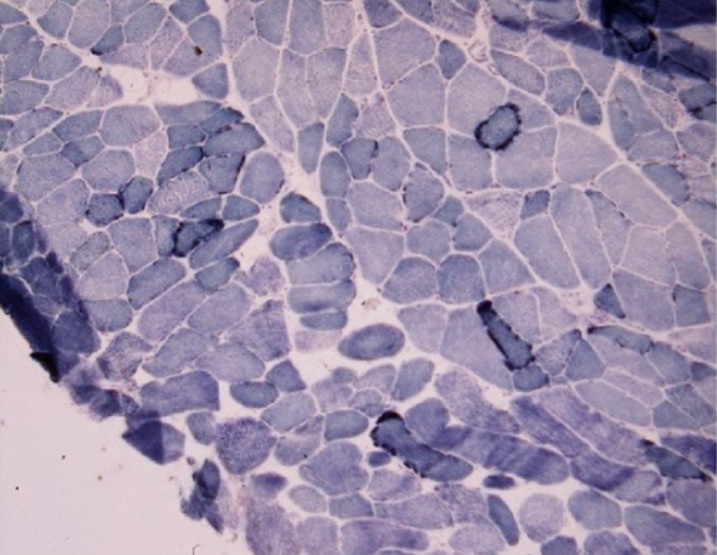
Muscle biopsy of a patient with myoclonus and progressive cognitive decline:
Ragged-blue fibers noted on succinate dehydrogenase preparation