Hypertrophic cardiomyopathy Cardiomyopathy Cardiomyopathy refers to a group of myocardial diseases associated with structural changes of the heart muscles (myocardium) and impaired systolic and/or diastolic function in the absence of other heart disorders (coronary artery disease, hypertension, valvular disease, and congenital heart disease). Cardiomyopathy: Overview and Types (HCM) is the most commonly inherited cardiomyopathy Cardiomyopathy Cardiomyopathy refers to a group of myocardial diseases associated with structural changes of the heart muscles (myocardium) and impaired systolic and/or diastolic function in the absence of other heart disorders (coronary artery disease, hypertension, valvular disease, and congenital heart disease). Cardiomyopathy: Overview and Types, which is characterized by an asymmetric increase in thickness ( hypertrophy Hypertrophy General increase in bulk of a part or organ due to cell enlargement and accumulation of fluids and secretions, not due to tumor formation, nor to an increase in the number of cells (hyperplasia). Cellular Adaptation) of the left ventricular wall, diastolic dysfunction Diastolic dysfunction Restrictive Cardiomyopathy, and often left ventricular outflow tract obstruction. Hypertrophic cardiomyopathy Cardiomyopathy Cardiomyopathy refers to a group of myocardial diseases associated with structural changes of the heart muscles (myocardium) and impaired systolic and/or diastolic function in the absence of other heart disorders (coronary artery disease, hypertension, valvular disease, and congenital heart disease). Cardiomyopathy: Overview and Types is caused by various gene Gene A category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms. Basic Terms of Genetics mutations affecting the contractile components of the heart, known as sarcomeres. Inheritance of HCM is typically autosomal dominant Autosomal dominant Autosomal inheritance, both dominant and recessive, refers to the transmission of genes from the 22 autosomal chromosomes. Autosomal dominant diseases are expressed when only 1 copy of the dominant allele is inherited. Autosomal Recessive and Autosomal Dominant Inheritance, although sporadic Sporadic Selective IgA Deficiency mutations also occur. Patients Patients Individuals participating in the health care system for the purpose of receiving therapeutic, diagnostic, or preventive procedures. Clinician–Patient Relationship may be asymptomatic, present with dyspnea Dyspnea Dyspnea is the subjective sensation of breathing discomfort. Dyspnea is a normal manifestation of heavy physical or psychological exertion, but also may be caused by underlying conditions (both pulmonary and extrapulmonary). Dyspnea and chest pain Pain An unpleasant sensation induced by noxious stimuli which are detected by nerve endings of nociceptive neurons. Pain: Types and Pathways or suffer sudden cardiac death Sudden cardiac death Cardiac arrest is the sudden, complete cessation of cardiac output with hemodynamic collapse. Patients present as pulseless, unresponsive, and apneic. Rhythms associated with cardiac arrest are ventricular fibrillation/tachycardia, asystole, or pulseless electrical activity. Cardiac Arrest without prior symptoms. Diagnosis is made based on ECG ECG An electrocardiogram (ECG) is a graphic representation of the electrical activity of the heart plotted against time. Adhesive electrodes are affixed to the skin surface allowing measurement of cardiac impulses from many angles. The ECG provides 3-dimensional information about the conduction system of the heart, the myocardium, and other cardiac structures. Electrocardiogram (ECG), echocardiography Echocardiography Ultrasonic recording of the size, motion, and composition of the heart and surrounding tissues. The standard approach is transthoracic. Tricuspid Valve Atresia (TVA), stress test, and cardiac MRI Cardiac MRI Imaging of the Heart and Great Vessels. Symptomatic HCM is typically treated with beta-blockers Beta-blockers Drugs that bind to but do not activate beta-adrenergic receptors thereby blocking the actions of beta-adrenergic agonists. Adrenergic beta-antagonists are used for treatment of hypertension, cardiac arrhythmias, angina pectoris, glaucoma, migraine headaches, and anxiety. Class 2 Antiarrhythmic Drugs (Beta Blockers) as the 1st-line therapy. Additional management depends on the presence of left ventricular outflow tract obstruction.
Last updated: Dec 15, 2025
Approximately 60%–70% of cases are caused by mutations affecting the thick or thin myofilament proteins Proteins Linear polypeptides that are synthesized on ribosomes and may be further modified, crosslinked, cleaved, or assembled into complex proteins with several subunits. The specific sequence of amino acids determines the shape the polypeptide will take, during protein folding, and the function of the protein. Energy Homeostasis of the sarcomeres (contractile components of the heart).
Many individuals affected by HCM are asymptomatic throughout their lives. For some, typically during adolescence, sudden cardiac death Sudden cardiac death Cardiac arrest is the sudden, complete cessation of cardiac output with hemodynamic collapse. Patients present as pulseless, unresponsive, and apneic. Rhythms associated with cardiac arrest are ventricular fibrillation/tachycardia, asystole, or pulseless electrical activity. Cardiac Arrest is the 1st symptom.
Symptoms:
Family history Family History Adult Health Maintenance:
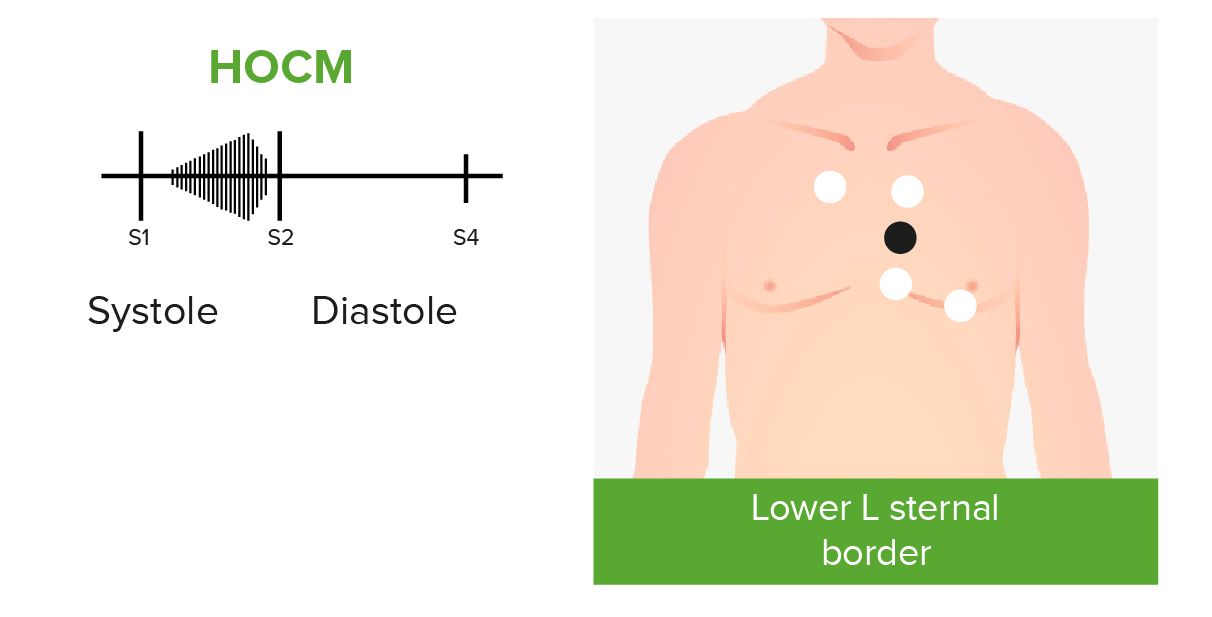
Phonocardiograms of abnormal heart sounds caused by hypertrophic obstructive cardiomyopathy (HOCM):
Patients with HOCM and left ventricular outflow tract obstruction may present with an audible crescendo-decrescendo systolic murmur best heard at the apex and lower left sternal border.
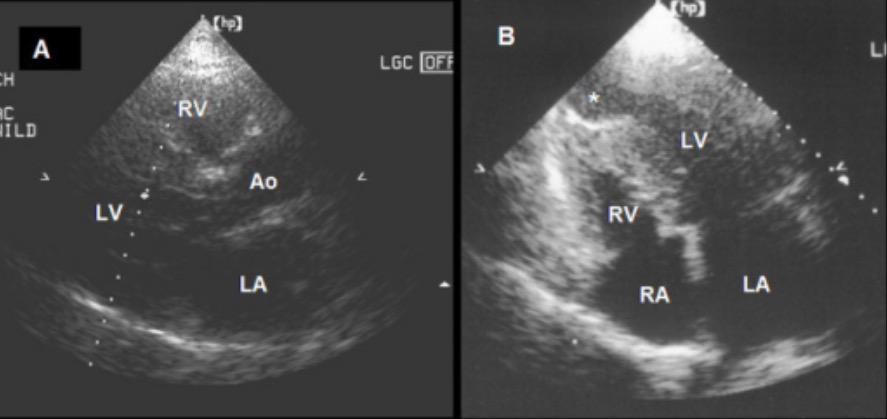
A: Parasternal long-axis view showing asymmetric septal hypertrophic cardiomyopathy in end diastole
B: A 4-chamber view showing a combination of basal and mid-ventricular hypertrophic cardiomyopathy and an apical aneurysm
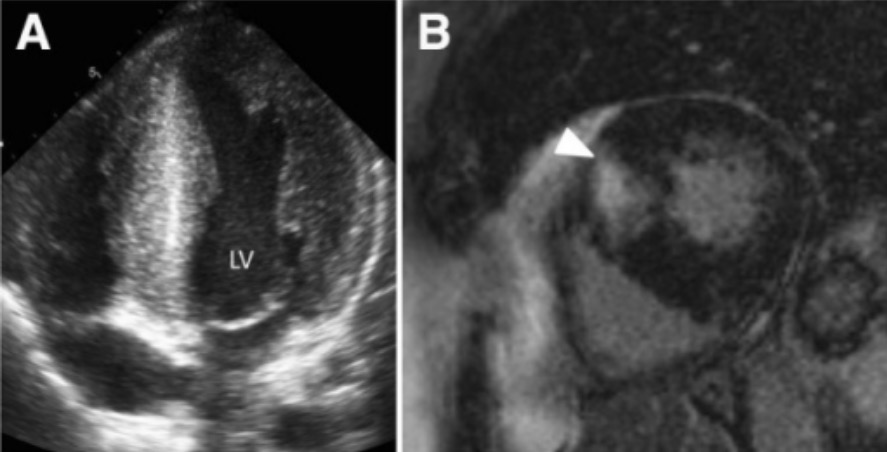
Hypertrophic cardiomyopathy: left ventricular (LV) hypertrophy with > 15-mm thickness of the septal and lateral walls (panel A)
Image: “Hypertrophic cardiomyopathy” by Victoria Delgado et al. License: CC BY 4.0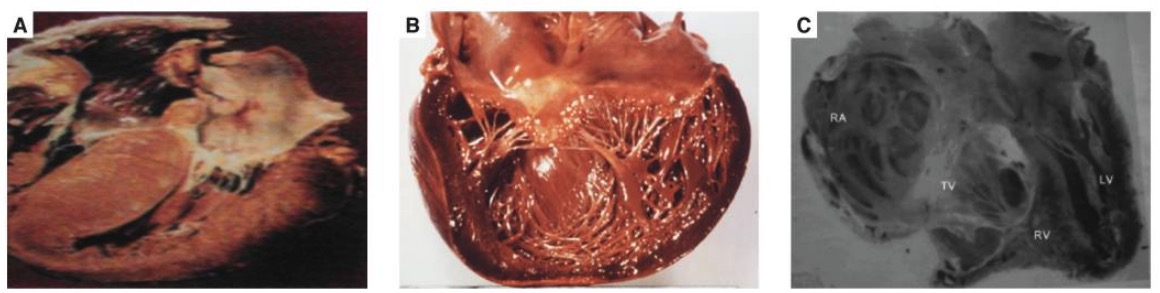
Hypertrophic cardiomyopathy
B: dilated cardiomyopathy
C: endomyocardial fibrosis
Goals of treatment:
Asymptomatic cases:
In setting of heart failure Heart Failure A heterogeneous condition in which the heart is unable to pump out sufficient blood to meet the metabolic need of the body. Heart failure can be caused by structural defects, functional abnormalities (ventricular dysfunction), or a sudden overload beyond its capacity. Chronic heart failure is more common than acute heart failure which results from sudden insult to cardiac function, such as myocardial infarction. Total Anomalous Pulmonary Venous Return (TAPVR) symptoms without left ventricular outflow tract obstruction:
In setting of heart failure Heart Failure A heterogeneous condition in which the heart is unable to pump out sufficient blood to meet the metabolic need of the body. Heart failure can be caused by structural defects, functional abnormalities (ventricular dysfunction), or a sudden overload beyond its capacity. Chronic heart failure is more common than acute heart failure which results from sudden insult to cardiac function, such as myocardial infarction. Total Anomalous Pulmonary Venous Return (TAPVR) symptoms and left ventricular outflow tract obstruction: