


As miopatias mitocondriais são patologias que surgem da disfunção das mitocôndrias (as estruturas produtoras de energia), caracterizadas por sintomas musculares proeminentes e acompanhadas por vários sintomas nos órgãos com elevadas necessidades energéticas. Os órgãos desproporcionalmente afetados incluem os músculos esqueléticos, o cérebro e o coração. As miopatias mitocondriais são causadas por mutações no DNA nuclear ou no DNA mitocondrial, que normalmente resultam na redução da produção de energia necessária pelas células. A apresentação pode ser uma miopatia isolada, encefalomiopatia, oftalmoplegia ou doença multissistémica. O diagnóstico envolve a colheita cuidada do historial médico e familiar, juntamente com estudos laboratoriais e genéticos. Na biopsia, há uma proliferação subsarcolémica e intermiofibrilar de mitocôndrias vistas como "fibras vermelhas irregulares". Esta situação clínica indica uma resposta compensatória à falha de energia. Não há um tratamento definitivo. O tratamento consiste em fisioterapia e numa abordagem multidisciplinar na resposta aos sintomas que acompanham a doença.
Last updated: Apr 24, 2025
As miopatias mitocondriais são doenças que surgem de disfunções das mitocôndrias (as estruturas produtoras de energia). Estas doenças são caracterizadas por sintomas musculares proeminentes (tais como fraqueza muscular) e acompanhadas por vários sintomas nos órgãos com elevadas necessidades energéticas.
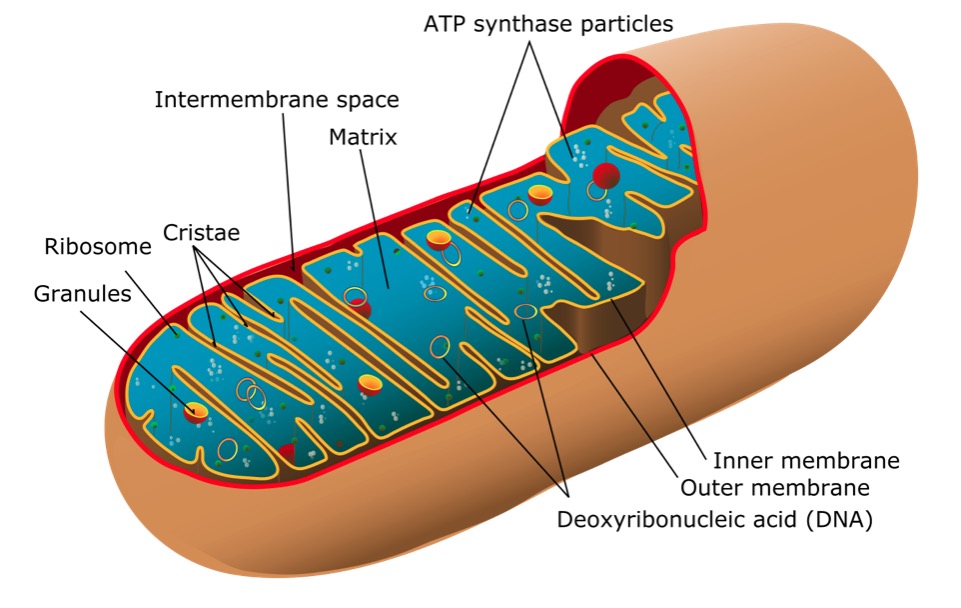
Estrutura de uma mitocôndria
Imagem: “Mitochondrion of the eukaryotic cell” por Mariana Ruiz Villarreal. Licença: Public DomainOs sinais e sintomas estão correlacionados com os órgãos ou tecidos afetados:

Síndrome de Kearns-Sayre (SKS):
Doente com ptose bilateral
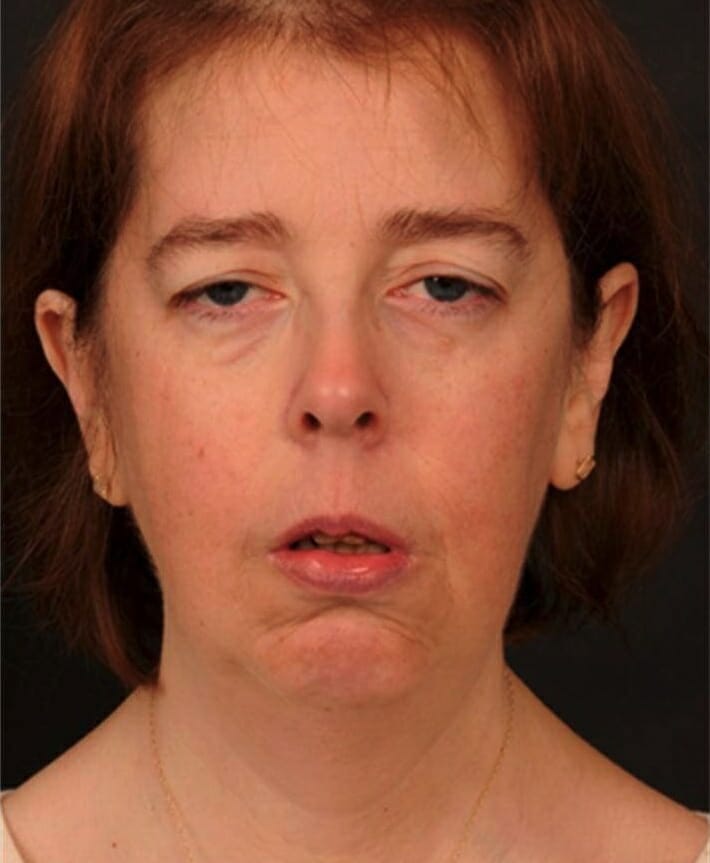
Características clínicas da mutação da epilepsia mioclónica e fibras vermelhas irregulares (MERRF, pela sigla em inglês): cabelo fino, ptose bilateral, biplegia facial com atrofia do músculo temporal, fraqueza do maxilar e fraqueza dos músculos flexor e extensor do pescoço
Imagem: “Clinical features of the MERRF mutation” por Wellcome Trust Centre for Mitochondrial Research, Institute for Ageing and Health, The Medical School, Newcastle University, Newcastle upon Tyne NE2 2HH, UK. Licença: CC BY 3.0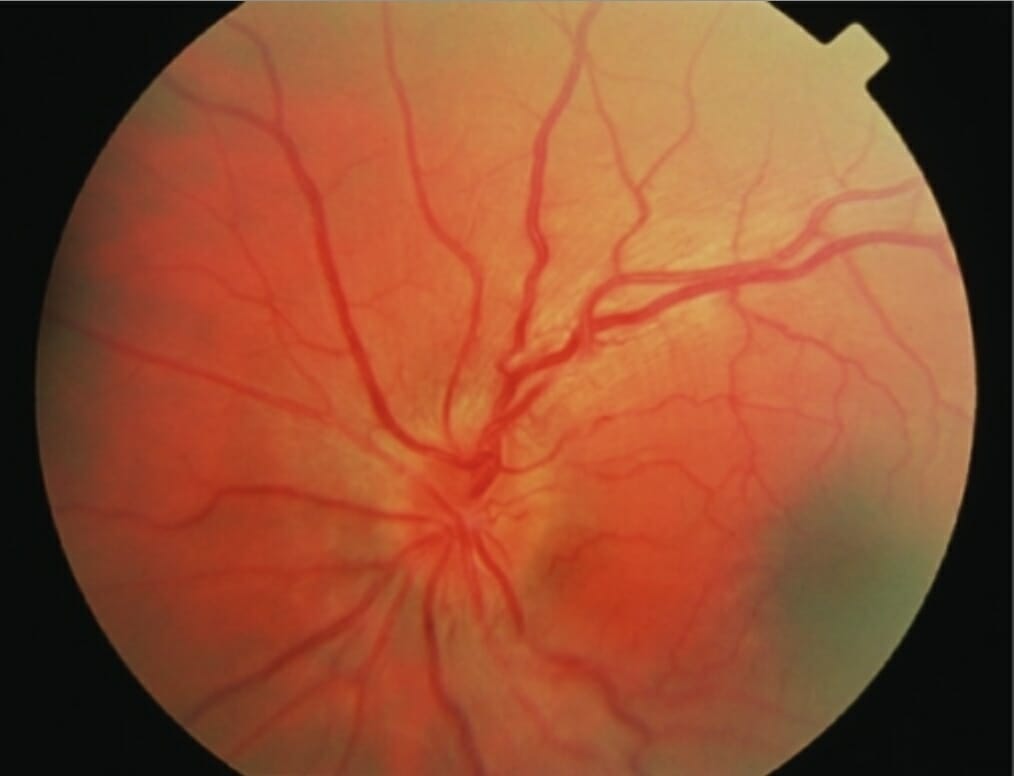
Neuropatia ótica hereditária de Leber com hiperémia do disco, tortuosidade da retina e edema da camada fibrosa do nervo retiniano peripapilar
Imagem: “Acute fundal appearance in Leber hereditary optic neuropathy” bporMitochondrial Research Group, The Medical School, Newcastle University, Newcastle upon Tyne, UK. Licença: CC BY 2.0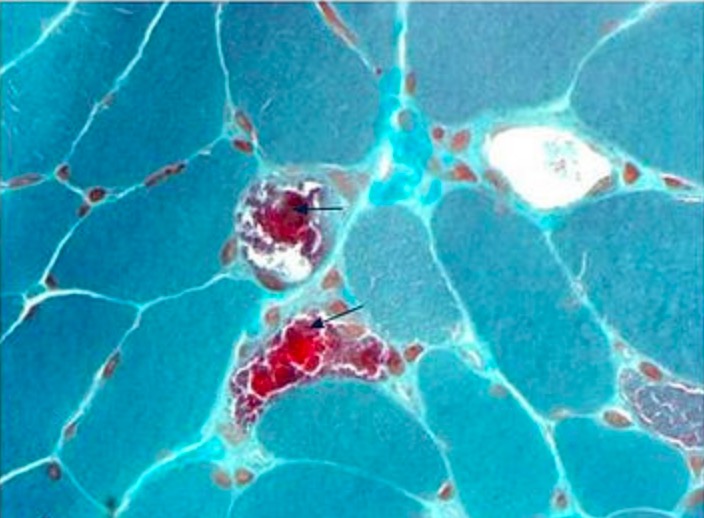
Biópsia muscular em paciente com características típicas de encefalomiopatia mitocondrial, acidose láctica e episódios tipo AVC:
Coloração tricrómica de Gomori modificada que mostra várias fibras vermelhas irregulares (setas)
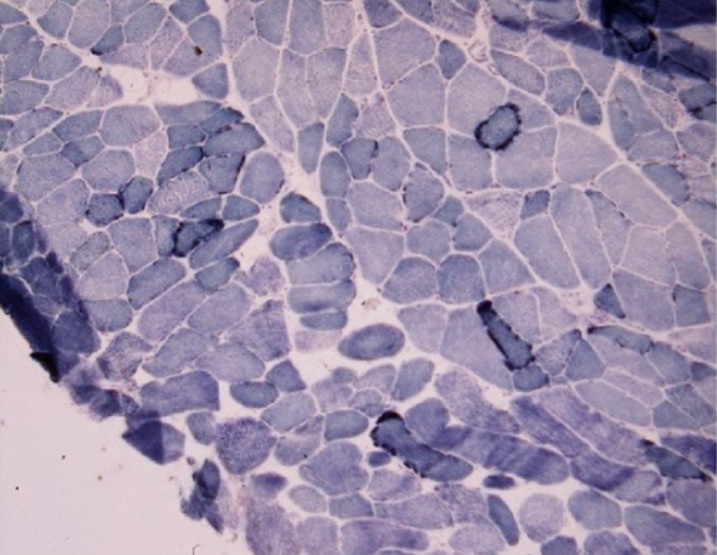
Biópsia muscular de um paciente com mioclonia e declínio cognitivo progressivo:
Fibras azuis irregulares observadas na preparação de succinato desidrogenase