Post-translational protein processing (including post-translational modification) is the folding, sorting, cleavage, and modifications required to make a protein functional after it is translated. As the protein folds, it forms complex secondary, tertiary, and quaternary structures. In addition, new functional groups or molecules may be added to the polypeptide chain, including phosphoryl, methyl, or acetyl groups; carbohydratesCarbohydratesA class of organic compounds composed of carbon, hydrogen, and oxygen in a ratio of cn(H2O)n. The largest class of organic compounds, including starch; glycogen; cellulose; polysaccharides; and simple monosaccharides.Basics of Carbohydrates; and lipidsLipidsLipids are a diverse group of hydrophobic organic molecules, which include fats, oils, sterols, and waxes.Fatty Acids and Lipids. ProteinsProteinsLinear polypeptides that are synthesized on ribosomes and may be further modified, crosslinked, cleaved, or assembled into complex proteins with several subunits. The specific sequence of amino acids determines the shape the polypeptide will take, during protein folding, and the function of the protein.Energy Homeostasis also have to be sorted into the correct intracellular compartment to either carry out their function, be packaged for secretionSecretionCoagulation Studies, or be inserted into the appropriate cellular membrane.
Amino acidsAmino acidsOrganic compounds that generally contain an amino (-NH2) and a carboxyl (-COOH) group. Twenty alpha-amino acids are the subunits which are polymerized to form proteins.Basics of Amino Acids are the building blocks of proteinsProteinsLinear polypeptides that are synthesized on ribosomes and may be further modified, crosslinked, cleaved, or assembled into complex proteins with several subunits. The specific sequence of amino acids determines the shape the polypeptide will take, during protein folding, and the function of the protein.Energy Homeostasis. Understanding the basics of amino acidsAmino acidsOrganic compounds that generally contain an amino (-NH2) and a carboxyl (-COOH) group. Twenty alpha-amino acids are the subunits which are polymerized to form proteins.Basics of Amino Acids allows a more comprehensive understanding of protein foldingProtein foldingProcesses involved in the formation of tertiary protein structure.Proteins and Peptides and modification.
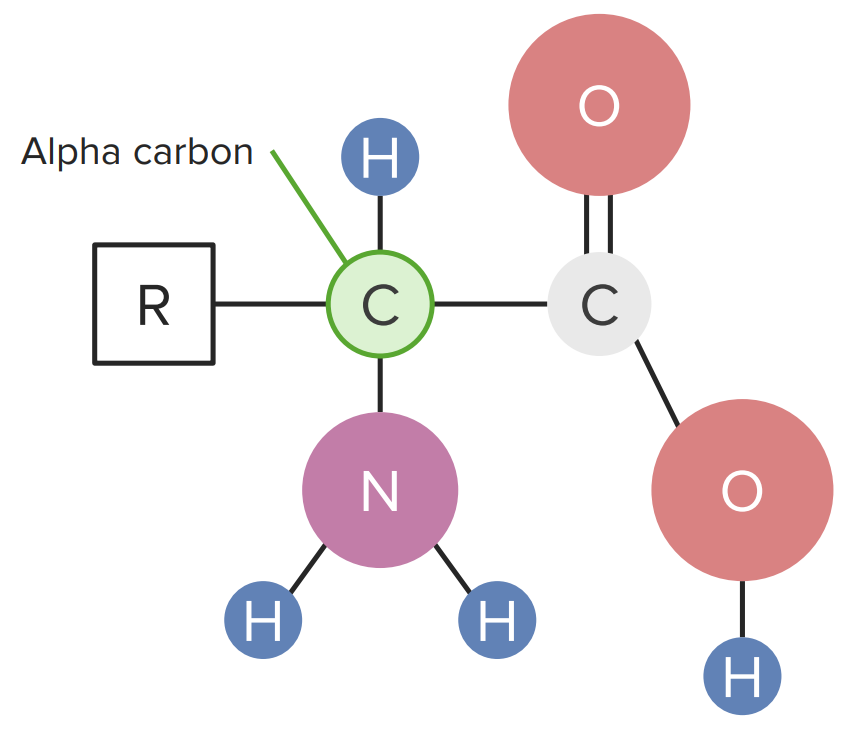
Amino acidAmino acidAmino acids (AAs) are composed of a central carbon atom attached to a carboxyl group, an amino group, a hydrogen atom, and a side chain (R group). Basics of Amino Acids structure
Amino acidsAmino acidsOrganic compounds that generally contain an amino (-NH2) and a carboxyl (-COOH) group. Twenty alpha-amino acids are the subunits which are polymerized to form proteins.Basics of Amino Acids that make up proteinsProteinsLinear polypeptides that are synthesized on ribosomes and may be further modified, crosslinked, cleaved, or assembled into complex proteins with several subunits. The specific sequence of amino acids determines the shape the polypeptide will take, during protein folding, and the function of the protein.Energy Homeostasis are known as α-amino acidsAcidsChemical compounds which yield hydrogen ions or protons when dissolved in water, whose hydrogen can be replaced by metals or basic radicals, or which react with bases to form salts and water (neutralization). An extension of the term includes substances dissolved in media other than water.Acid-Base Balance. Each of these amino acidsAmino acidsOrganic compounds that generally contain an amino (-NH2) and a carboxyl (-COOH) group. Twenty alpha-amino acids are the subunits which are polymerized to form proteins.Basics of Amino Acids has a central carbon known as the “alpha carbon,” which makes 4 bonds:
Hydrogen ion
Carboxyl group: made up of carboxylic acid (–COOH), creating the C-terminal end of the amino acidAmino acidAmino acids (AAs) are composed of a central carbon atom attached to a carboxyl group, an amino group, a hydrogen atom, and a side chain (R group). Basics of Amino Acids
Amine group: made up of an amine group (–NH2), creating the N-terminal end of the amino acidAmino acidAmino acids (AAs) are composed of a central carbon atom attached to a carboxyl group, an amino group, a hydrogen atom, and a side chain (R group). Basics of Amino Acids
R side chain: the unique functional group of an amino acidAmino acidAmino acids (AAs) are composed of a central carbon atom attached to a carboxyl group, an amino group, a hydrogen atom, and a side chain (R group). Basics of Amino Acids
Diagram of an amino acid
Image by Lecturio.
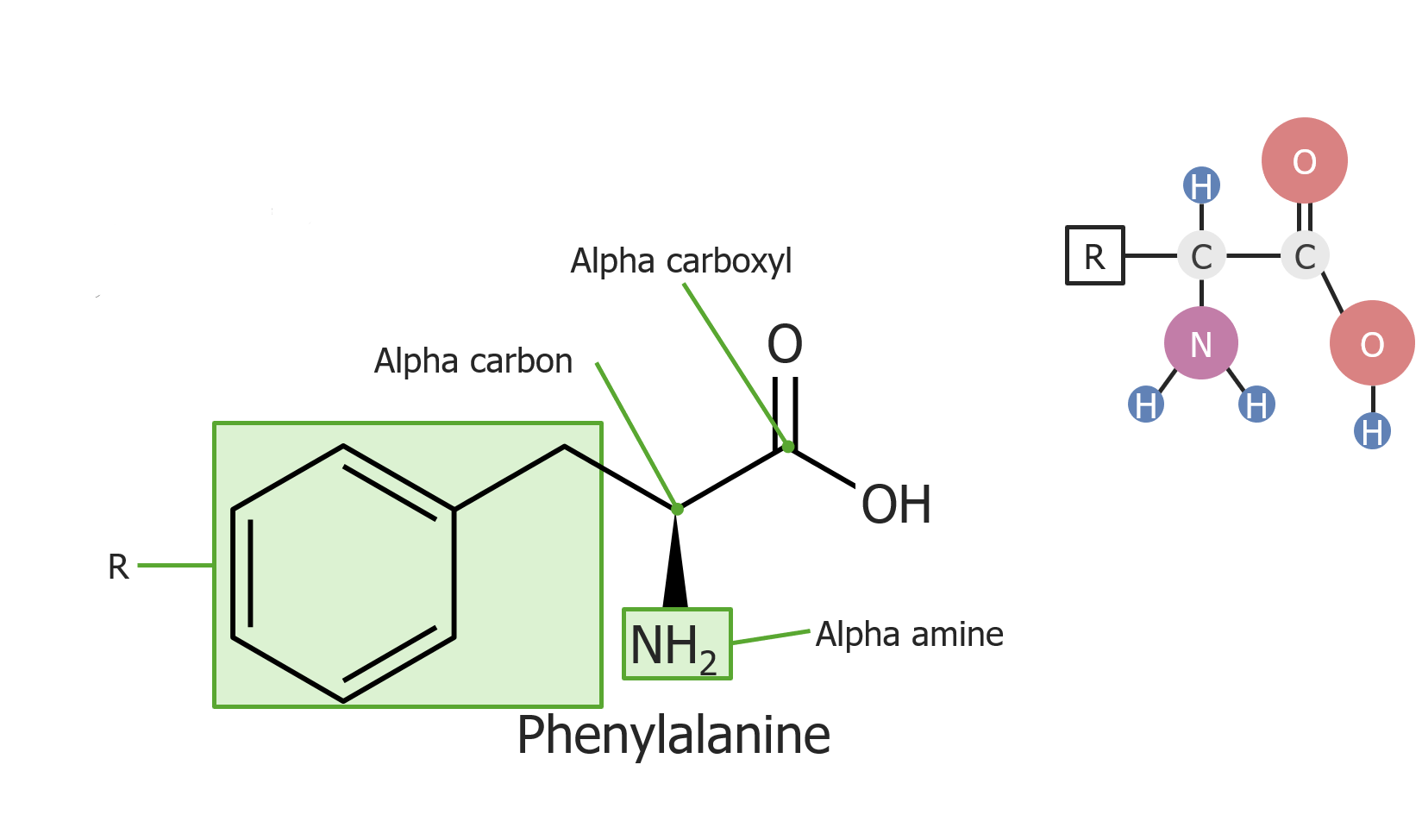
Example of the amino acid phenylalanine
Image by Lecturio.
Properties of amino acidsAmino acidsOrganic compounds that generally contain an amino (-NH2) and a carboxyl (-COOH) group. Twenty alpha-amino acids are the subunits which are polymerized to form proteins.Basics of Amino Acids
Amino acidsAmino acidsOrganic compounds that generally contain an amino (-NH2) and a carboxyl (-COOH) group. Twenty alpha-amino acids are the subunits which are polymerized to form proteins.Basics of Amino Acids may be categorized by characteristics of their R groups, which may be:
Protein structure, which is often referred to as protein foldingProtein foldingProcesses involved in the formation of tertiary protein structure.Proteins and Peptides, has 4 levels. These levels are:
Primary
Secondary
Tertiary
Quaternary
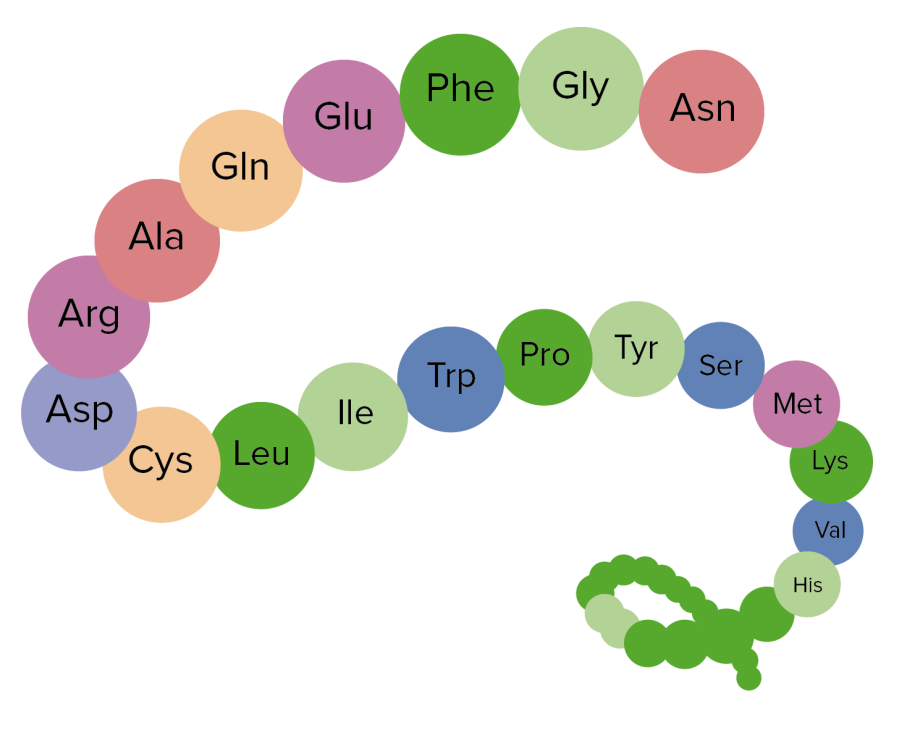
Primary structure
The primary structure is the linear sequence of the amino acidsAmino acidsOrganic compounds that generally contain an amino (-NH2) and a carboxyl (-COOH) group. Twenty alpha-amino acids are the subunits which are polymerized to form proteins.Basics of Amino Acids in the peptide chain.
“Beads on a string” joined by peptide bonds
This structure ultimately determines all the properties of the protein.
Example of primary structure of a protein
Image by Lecturio.
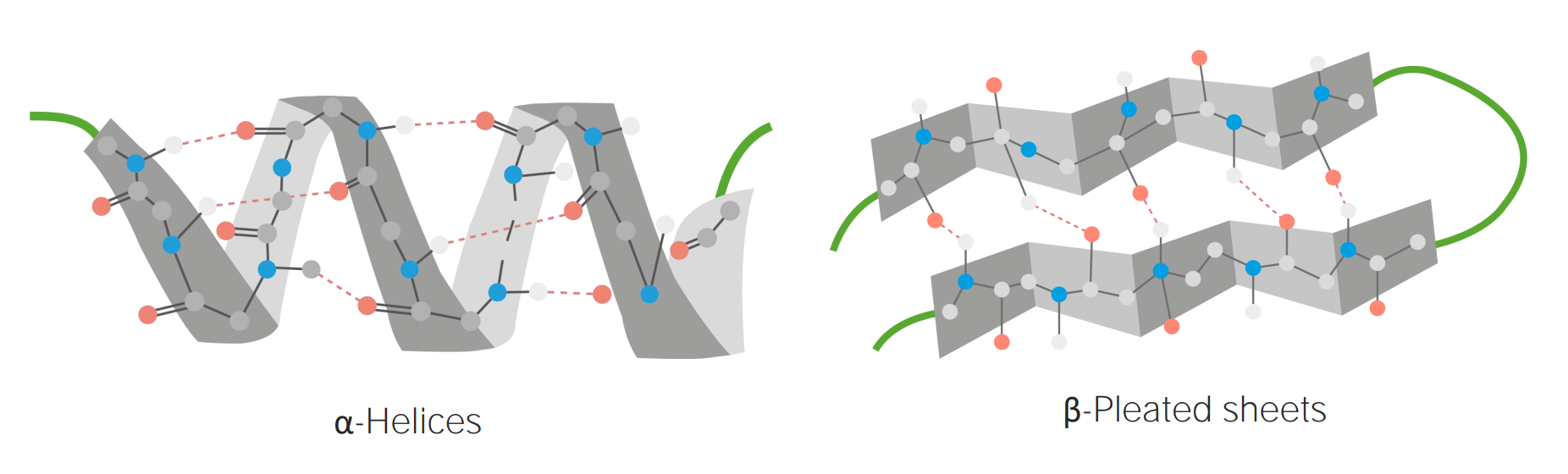
Secondary structure
Occurs between amino acidsAmino acidsOrganic compounds that generally contain an amino (-NH2) and a carboxyl (-COOH) group. Twenty alpha-amino acids are the subunits which are polymerized to form proteins.Basics of Amino Acids that are relatively close to each other (typically about 3–10 amino acidsAmino acidsOrganic compounds that generally contain an amino (-NH2) and a carboxyl (-COOH) group. Twenty alpha-amino acids are the subunits which are polymerized to form proteins.Basics of Amino Acids apart)
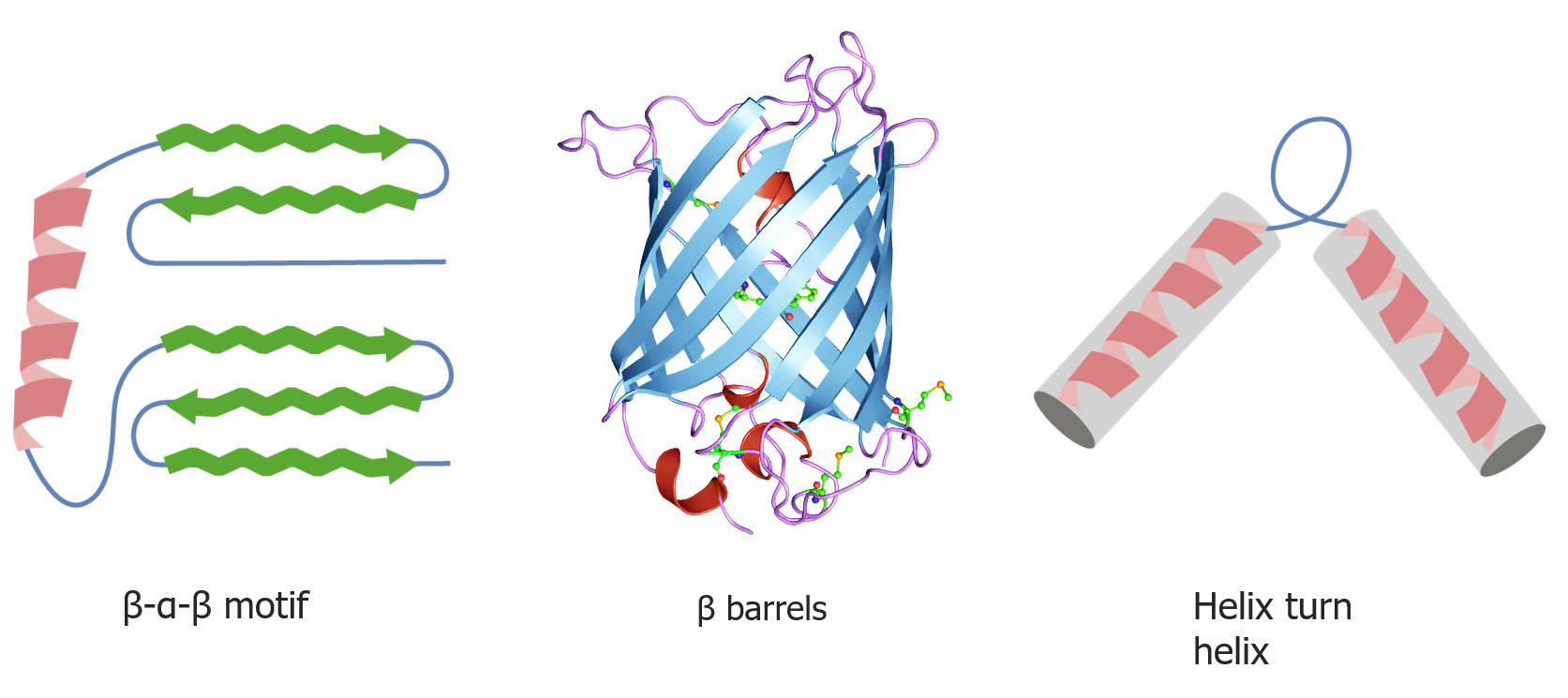
3 common motifs include:
α-helix: a coil with the R groups on the outside
β-strands and sheets:
planar structure formed by zigzagging amino acidAmino acidAmino acids (AAs) are composed of a central carbon atom attached to a carboxyl group, an amino group, a hydrogen atom, and a side chain (R group). Basics of Amino Acids strands
R groups protrude from the top and bottom of the sheet.
Reverse turns: a short sequence, usually involving prolineProlineA non-essential amino acid that is synthesized from glutamic acid. It is an essential component of collagen and is important for proper functioning of joints and tendons.Synthesis of Nonessential Amino Acids and/or glycineGlycineA non-essential amino acid. It is found primarily in gelatin and silk fibroin and used therapeutically as a nutrient. It is also a fast inhibitory neurotransmitter.Synthesis of Nonessential Amino Acids, occurring between α-helices and/or β-pleated sheets
Secondary structures are stabilized by hydrogen bonds between the carboxyl oxygen and the amine hydrogens.
Some simple fibrousFibrousFibrocystic ChangeproteinsProteinsLinear polypeptides that are synthesized on ribosomes and may be further modified, crosslinked, cleaved, or assembled into complex proteins with several subunits. The specific sequence of amino acids determines the shape the polypeptide will take, during protein folding, and the function of the protein.Energy Homeostasis (e.g., keratinKeratinA class of fibrous proteins or scleroproteins that represents the principal constituent of epidermis; hair; nails; horny tissues, and the organic matrix of tooth enamel. Two major conformational groups have been characterized, alpha-keratin, whose peptide backbone forms a coiled-coil alpha helical structure consisting of type I keratin and a type II keratin, and beta-keratin, whose backbone forms a zigzag or pleated sheet structure. Alpha-keratins have been classified into at least 20 subtypes. In addition multiple isoforms of subtypes have been found which may be due to gene duplication.Seborrheic Keratosis) have only primary and secondary structures.
Computer models are often able to predict secondary structures based on the amino acidAmino acidAmino acids (AAs) are composed of a central carbon atom attached to a carboxyl group, an amino group, a hydrogen atom, and a side chain (R group). Basics of Amino Acids sequence.
Examples of α-helices and β-pleated sheets
Image by Lecturio.
Tertiary structure
Tertiary structure is the complex looping and folding that occurs as a result of interactions and bonding between portions of the protein that are farther apart. Examples of interactions that create tertiary structure include:
Hydrogen bonds: form between polar side chains
Disulfide bridges: strong covalent bonds that form between two cysteines
Ionic bonds: form between a positively charged/acidic R group (e.g., carboxyl group on aspartic acid) and a negatively charged/basic R group (e.g., amine group on lysine)
Metallic bonds: 2 regions of a protein bound to a metal (e.g., ironIronA metallic element with atomic symbol fe, atomic number 26, and atomic weight 55. 85. It is an essential constituent of hemoglobins; cytochromes; and iron-binding proteins. It plays a role in cellular redox reactions and in the transport of oxygen.Trace Elements)
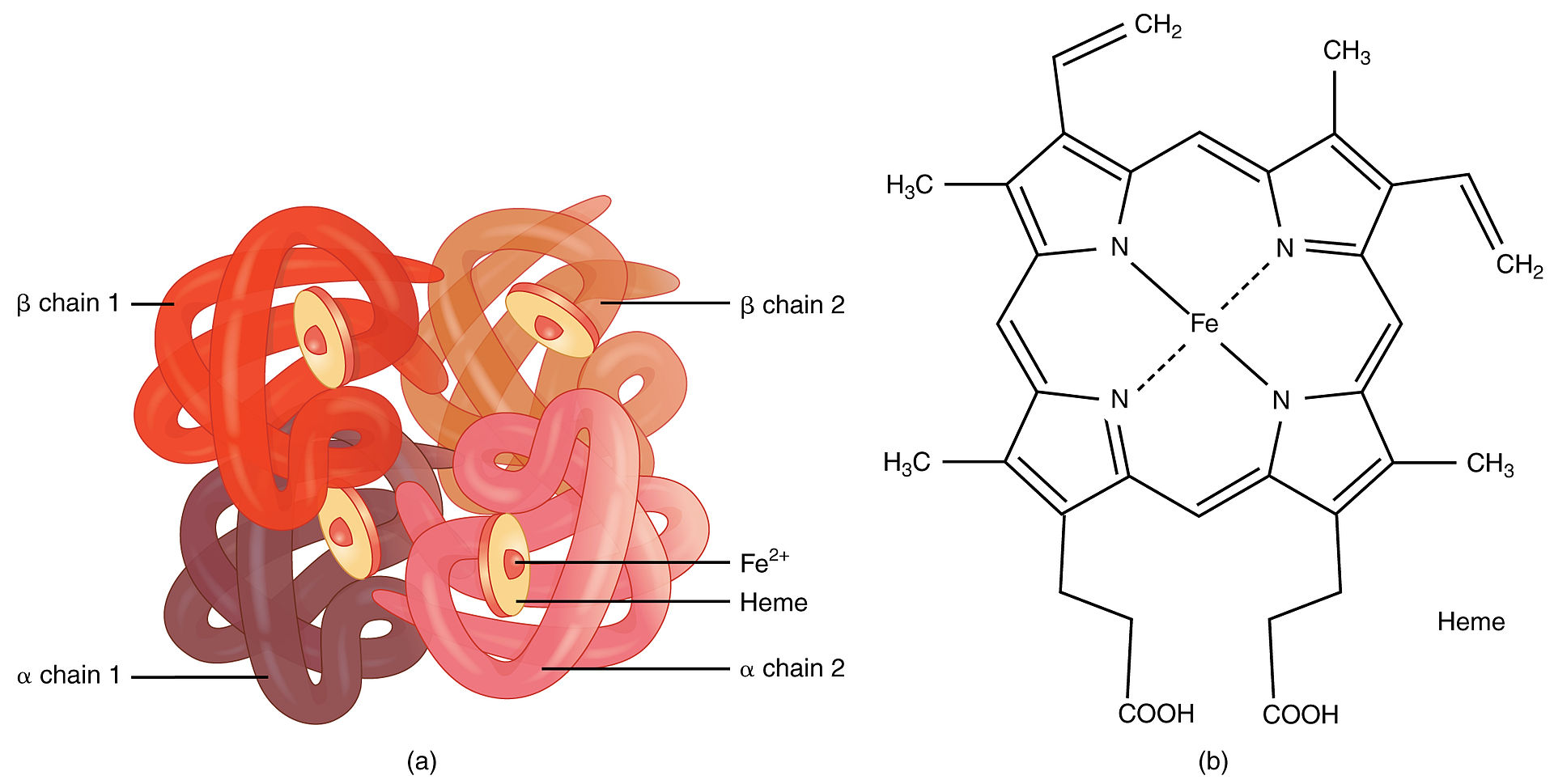
An example of quaternary structure Image: “1904 Hemoglobin” by OpenStax College. License: CC BY 3.0
Quaternary and tertiary protein folding motifs
Image by Lecturio.
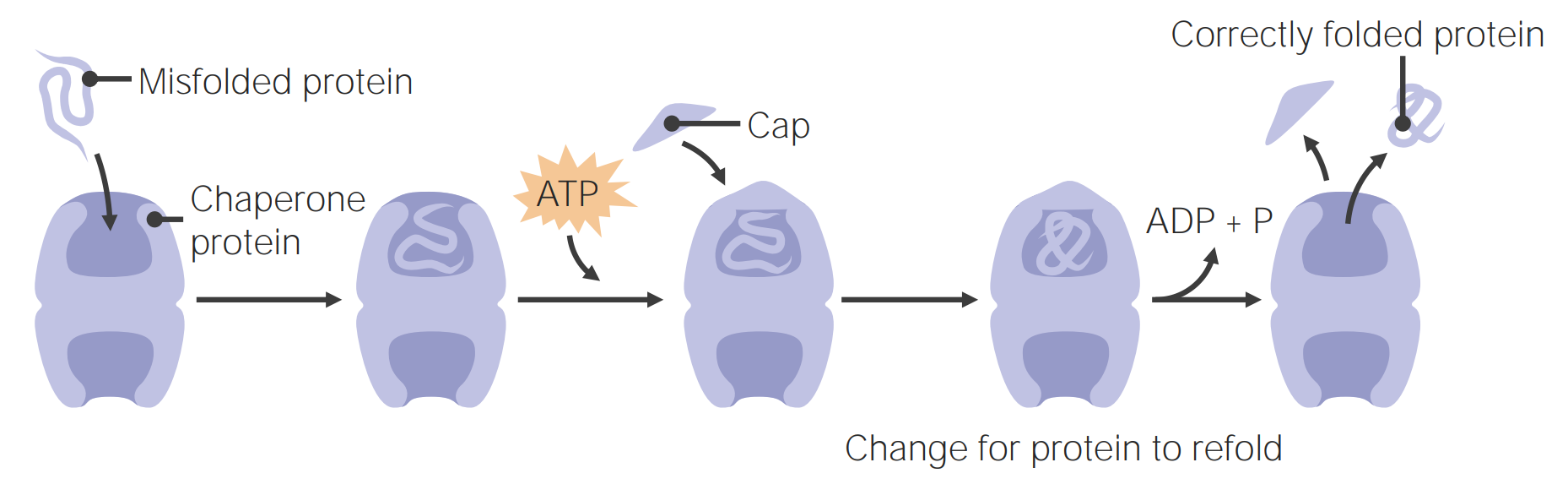
Chaperone proteinsProteinsLinear polypeptides that are synthesized on ribosomes and may be further modified, crosslinked, cleaved, or assembled into complex proteins with several subunits. The specific sequence of amino acids determines the shape the polypeptide will take, during protein folding, and the function of the protein.Energy Homeostasis
Chaperone proteinsProteinsLinear polypeptides that are synthesized on ribosomes and may be further modified, crosslinked, cleaved, or assembled into complex proteins with several subunits. The specific sequence of amino acids determines the shape the polypeptide will take, during protein folding, and the function of the protein.Energy Homeostasis assist in protein foldingProtein foldingProcesses involved in the formation of tertiary protein structure.Proteins and Peptides.
ChaperonesChaperonesA family of cellular proteins that mediate the correct assembly or disassembly of polypeptides and their associated ligands. Although they take part in the assembly process, molecular chaperones are not components of the final structures.Cell Injury and Deathare barrel-like proteinsProteinsLinear polypeptides that are synthesized on ribosomes and may be further modified, crosslinked, cleaved, or assembled into complex proteins with several subunits. The specific sequence of amino acids determines the shape the polypeptide will take, during protein folding, and the function of the protein.Energy Homeostasis that take in misfolded proteinsMisfolded ProteinsCell Injury and Death and use ATP energy to refold them.
These proteinsProteinsLinear polypeptides that are synthesized on ribosomes and may be further modified, crosslinked, cleaved, or assembled into complex proteins with several subunits. The specific sequence of amino acids determines the shape the polypeptide will take, during protein folding, and the function of the protein.Energy Homeostasis can bindBINDHyperbilirubinemia of the Newborn to hydrophobic regions of unfolded proteinsProteinsLinear polypeptides that are synthesized on ribosomes and may be further modified, crosslinked, cleaved, or assembled into complex proteins with several subunits. The specific sequence of amino acids determines the shape the polypeptide will take, during protein folding, and the function of the protein.Energy Homeostasis, allowing proper folding to take place.
Found in various cellular compartments such as:
CytosolCytosolA cell’s cytoskeleton is a network of intracellular protein fibers that provides structural support, anchors organelles, and aids intra- and extracellular movement.The Cell: Cytosol and Cytoskeleton
MitochondriaMitochondriaSemiautonomous, self-reproducing organelles that occur in the cytoplasm of all cells of most, but not all, eukaryotes. Each mitochondrion is surrounded by a double limiting membrane. The inner membrane is highly invaginated, and its projections are called cristae. Mitochondria are the sites of the reactions of oxidative phosphorylation, which result in the formation of ATP. They contain distinctive ribosomes, transfer RNAs; amino Acyl tRNA synthetases; and elongation and termination factors. Mitochondria depend upon genes within the nucleus of the cells in which they reside for many essential messenger RNAs. Mitochondria are believed to have arisen from aerobic bacteria that established a symbiotic relationship with primitive protoeukaryotes.The Cell: Organelles
Lumen of the endoplasmic reticulumEndoplasmic reticulumA system of cisternae in the cytoplasm of many cells. In places the endoplasmic reticulum is continuous with the plasma membrane (cell membrane) or outer membrane of the nuclear envelope. If the outer surfaces of the endoplasmic reticulum membranes are coated with ribosomes, the endoplasmic reticulum is said to be rough-surfaced; otherwise it is said to be smooth-surfaced.The Cell: Organelles
Chaperone proteins assist in protein folding
Image by Lecturio.
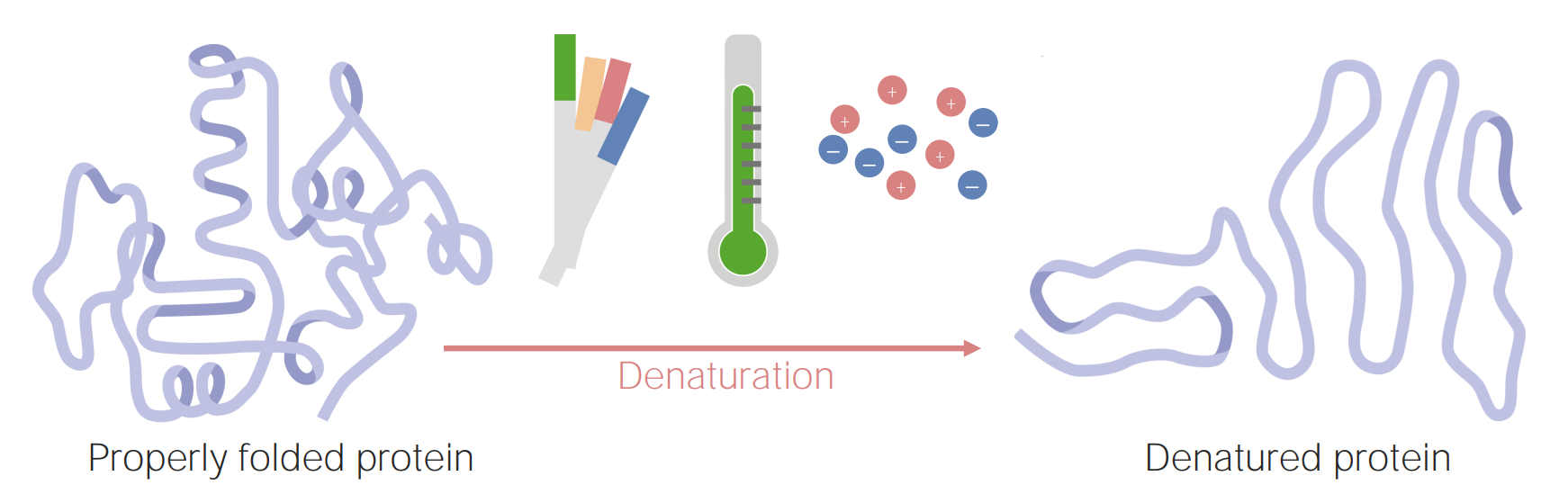
Protein denaturing
A denatured protein is a protein that has been unfoldedand is no longer functional. This unfolding occurs under certain conditions, which include changes in:
pHpHThe quantitative measurement of the acidity or basicity of a solution.Acid-Base Balance
Temperature
Ionic concentration
Proteins can become denatured (or unfolded) as a result of changes in pH, temperature, or ionic concentration.
ProteinsProteinsLinear polypeptides that are synthesized on ribosomes and may be further modified, crosslinked, cleaved, or assembled into complex proteins with several subunits. The specific sequence of amino acids determines the shape the polypeptide will take, during protein folding, and the function of the protein.Energy Homeostasis need to be sorted and will end up remaining in the cell, being placed on the cell wallCell wallThe outermost layer of a cell in most plants; bacteria; fungi; and algae. The cell wall is usually a rigid structure that lies external to the cell membrane, and provides a protective barrier against physical or chemical agents.Cell Types: Eukaryotic versus Prokaryotic, or being exported/secreted.
Exported and surface proteinsProteinsLinear polypeptides that are synthesized on ribosomes and may be further modified, crosslinked, cleaved, or assembled into complex proteins with several subunits. The specific sequence of amino acids determines the shape the polypeptide will take, during protein folding, and the function of the protein.Energy Homeostasis
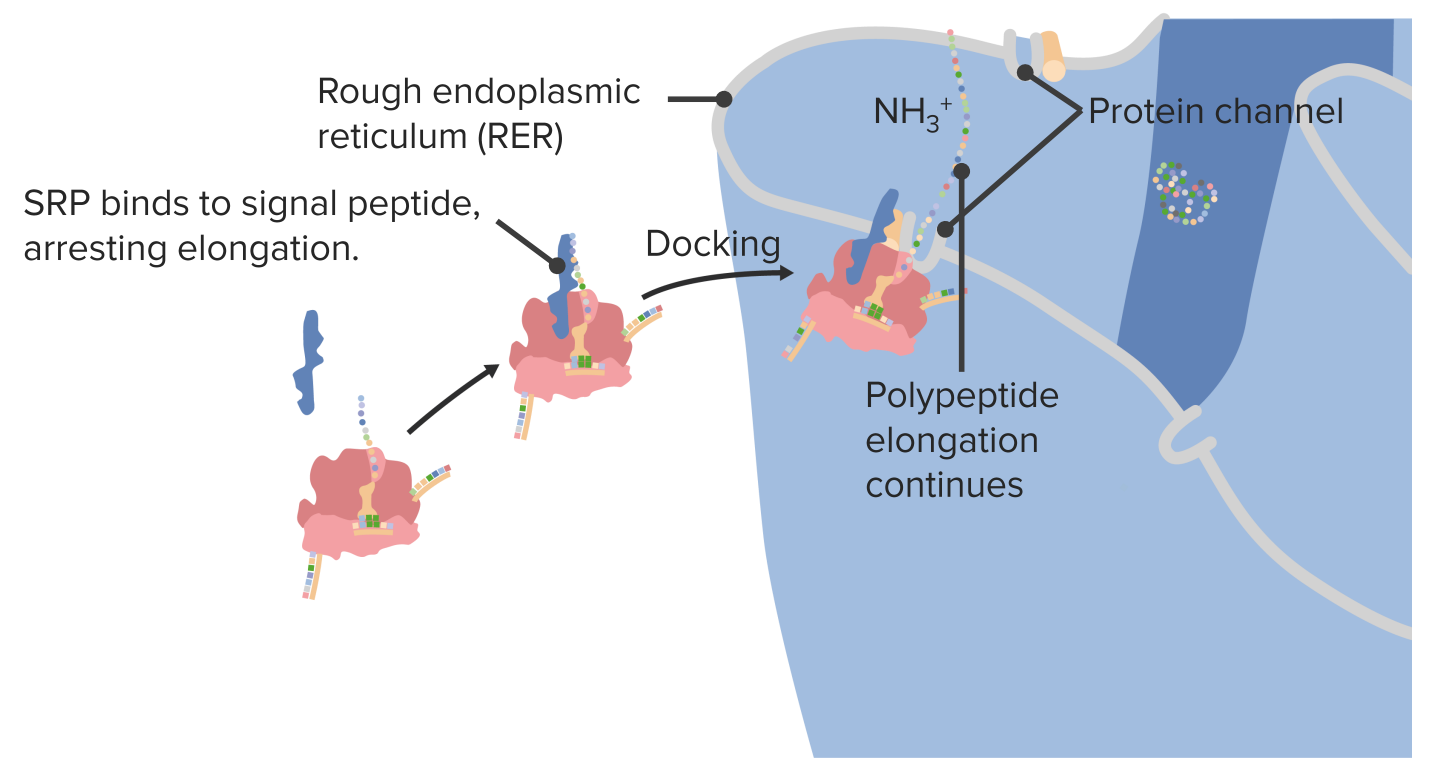
ProteinsProteinsLinear polypeptides that are synthesized on ribosomes and may be further modified, crosslinked, cleaved, or assembled into complex proteins with several subunits. The specific sequence of amino acids determines the shape the polypeptide will take, during protein folding, and the function of the protein.Energy Homeostasis destined for the cell surface and/or secretionSecretionCoagulation Studies from the cell are synthesized within the rough endoplasmic reticulumEndoplasmic reticulumA system of cisternae in the cytoplasm of many cells. In places the endoplasmic reticulum is continuous with the plasma membrane (cell membrane) or outer membrane of the nuclear envelope. If the outer surfaces of the endoplasmic reticulum membranes are coated with ribosomes, the endoplasmic reticulum is said to be rough-surfaced; otherwise it is said to be smooth-surfaced.The Cell: Organelles (RER):
During translationTranslationTranslation is the process of synthesizing a protein from a messenger RNA (mRNA) transcript. This process is divided into three primary stages: initiation, elongation, and termination. Translation is catalyzed by structures known as ribosomes, which are large complexes of proteins and ribosomal RNA (rRNA). Stages and Regulation of Translation, a signal peptide sequence on the end of the growing polypeptide chain indicates that a protein is destined for secretionSecretionCoagulation Studies from the cell.
A signal recognition protein (SRPSRPRefers to the presence of the phenomenon in association with an underlying illness, most commonly an autoimmune disease such as systemic sclerosis or systemic lupus erythematosus; or with drug use, from occupational hazards, etcRaynaud Phenomenon) binds to the signal peptide sequence, pausing elongationElongationPolymerase Chain Reaction (PCR).
The SRPSRPRefers to the presence of the phenomenon in association with an underlying illness, most commonly an autoimmune disease such as systemic sclerosis or systemic lupus erythematosus; or with drug use, from occupational hazards, etcRaynaud Phenomenon guides the entire ribosome to the RER and associates it with a pore.
ProteinsProteinsLinear polypeptides that are synthesized on ribosomes and may be further modified, crosslinked, cleaved, or assembled into complex proteins with several subunits. The specific sequence of amino acids determines the shape the polypeptide will take, during protein folding, and the function of the protein.Energy Homeostasis are then directed to the lysosomesLysosomesA class of morphologically heterogeneous cytoplasmic particles in animal and plant tissues characterized by their content of hydrolytic enzymes and the structure-linked latency of these enzymes. The intracellular functions of lysosomes depend on their lytic potential. The single unit membrane of the lysosome acts as a barrier between the enzymes enclosed in the lysosome and the external substrate. The activity of the enzymes contained in lysosomes is limited or nil unless the vesicle in which they are enclosed is ruptured or undergoes membrane fusion.The Cell: Organelles or plasma membranePlasma membraneA cell membrane (also known as the plasma membrane or plasmalemma) is a biological membrane that separates the cell contents from the outside environment. A cell membrane is composed of a phospholipid bilayer and proteins that function to protect cellular DNA and mediate the exchange of ions and molecules.The Cell: Cell Membrane or packaged for exocytosisExocytosisCellular release of material within membrane-limited vesicles by fusion of the vesicles with the cell membrane.The Cell: Cell Membrane (secretionSecretionCoagulation Studies):
Secreted proteinsProteinsLinear polypeptides that are synthesized on ribosomes and may be further modified, crosslinked, cleaved, or assembled into complex proteins with several subunits. The specific sequence of amino acids determines the shape the polypeptide will take, during protein folding, and the function of the protein.Energy Homeostasis follow the exocytic (secretory) pathway: RER → Golgi apparatus (GA) → plasma membranePlasma membraneA cell membrane (also known as the plasma membrane or plasmalemma) is a biological membrane that separates the cell contents from the outside environment. A cell membrane is composed of a phospholipid bilayer and proteins that function to protect cellular DNA and mediate the exchange of ions and molecules.The Cell: Cell Membrane (PM)
ProteinsProteinsLinear polypeptides that are synthesized on ribosomes and may be further modified, crosslinked, cleaved, or assembled into complex proteins with several subunits. The specific sequence of amino acids determines the shape the polypeptide will take, during protein folding, and the function of the protein.Energy Homeostasis destined for the GA, PM, or secretionSecretionCoagulation Studies are carried in transport vesiclesVesiclesFemale Genitourinary Examination.
Docking a ribosome on the rough endoplasmic reticulum SRP: signal recognition protein
Image by Lecturio.
Intracellular proteinsProteinsLinear polypeptides that are synthesized on ribosomes and may be further modified, crosslinked, cleaved, or assembled into complex proteins with several subunits. The specific sequence of amino acids determines the shape the polypeptide will take, during protein folding, and the function of the protein.Energy Homeostasis
ProteinsProteinsLinear polypeptides that are synthesized on ribosomes and may be further modified, crosslinked, cleaved, or assembled into complex proteins with several subunits. The specific sequence of amino acids determines the shape the polypeptide will take, during protein folding, and the function of the protein.Energy Homeostasis synthesized in the cytosolCytosolA cell’s cytoskeleton is a network of intracellular protein fibers that provides structural support, anchors organelles, and aids intra- and extracellular movement.The Cell: Cytosol and Cytoskeleton, unassociated with the RER, are kept inside the cell.
Other specific signal peptides may direct these proteinsProteinsLinear polypeptides that are synthesized on ribosomes and may be further modified, crosslinked, cleaved, or assembled into complex proteins with several subunits. The specific sequence of amino acids determines the shape the polypeptide will take, during protein folding, and the function of the protein.Energy Homeostasis to their final location in the cell (e.g., the nucleusNucleusWithin a eukaryotic cell, a membrane-limited body which contains chromosomes and one or more nucleoli (cell nucleolus). The nuclear membrane consists of a double unit-type membrane which is perforated by a number of pores; the outermost membrane is continuous with the endoplasmic reticulum. A cell may contain more than one nucleus.The Cell: Organelles).
After a polypeptide is synthesized, it undergoes further modification in order to form a functional protein. This modification may include cleaving off portions of the polypeptide chain or adding a functional group.
Protein cleavage
Protein cleavage is the process of removing certain polypeptides in order for the protein to become functional.
Many proteinsProteinsLinear polypeptides that are synthesized on ribosomes and may be further modified, crosslinked, cleaved, or assembled into complex proteins with several subunits. The specific sequence of amino acids determines the shape the polypeptide will take, during protein folding, and the function of the protein.Energy Homeostasis are not functional immediately after translationTranslationTranslation is the process of synthesizing a protein from a messenger RNA (mRNA) transcript. This process is divided into three primary stages: initiation, elongation, and termination. Translation is catalyzed by structures known as ribosomes, which are large complexes of proteins and ribosomal RNA (rRNA). Stages and Regulation of Translation; these proteinsProteinsLinear polypeptides that are synthesized on ribosomes and may be further modified, crosslinked, cleaved, or assembled into complex proteins with several subunits. The specific sequence of amino acids determines the shape the polypeptide will take, during protein folding, and the function of the protein.Energy Homeostasis are called pro-proteins.
Examples of amino acidsAmino acidsOrganic compounds that generally contain an amino (-NH2) and a carboxyl (-COOH) group. Twenty alpha-amino acids are the subunits which are polymerized to form proteins.Basics of Amino Acids or peptides that are typically cleaved off:
The first amino acidAmino acidAmino acids (AAs) are composed of a central carbon atom attached to a carboxyl group, an amino group, a hydrogen atom, and a side chain (R group). Basics of Amino Acids (methionine) matches the start codonCodonA set of three nucleotides in a protein coding sequence that specifies individual amino acids or a termination signal. Most codons are universal, but some organisms do not produce the transfer RNAs complementary to all codons. These codons are referred to as unassigned codons.Basic Terms of Genetics in mRNAmRNARNA sequences that serve as templates for protein synthesis. Bacterial mRNAs are generally primary transcripts in that they do not require post-transcriptional processing. Eukaryotic mRNA is synthesized in the nucleus and must be exported to the cytoplasm for translation. Most eukaryotic mRNAs have a sequence of polyadenylic acid at the 3′ end, referred to as the poly(a) tail. The function of this tail is not known for certain, but it may play a role in the export of mature mRNA from the nucleus as well as in helping stabilize some mRNA molecules by retarding their degradation in the cytoplasm.RNA Types and Structure, signaling the start of translationTranslationTranslation is the process of synthesizing a protein from a messenger RNA (mRNA) transcript. This process is divided into three primary stages: initiation, elongation, and termination. Translation is catalyzed by structures known as ribosomes, which are large complexes of proteins and ribosomal RNA (rRNA). Stages and Regulation of Translation.
Signal peptides assist the protein in getting to its proper location but are not part of the functional protein itself.
EnzymesEnzymesEnzymes are complex protein biocatalysts that accelerate chemical reactions without being consumed by them. Due to the body’s constant metabolic needs, the absence of enzymes would make life unsustainable, as reactions would occur too slowly without these molecules. Basics of Enzymes and hormonesHormonesHormones are messenger molecules that are synthesized in one part of the body and move through the bloodstream to exert specific regulatory effects on another part of the body. Hormones play critical roles in coordinating cellular activities throughout the body in response to the constant changes in both the internal and external environments. Hormones: Overview and Types are frequently translated as pro-proteins, which require cleavage in order to become functional/active (e.g., insulinInsulinInsulin is a peptide hormone that is produced by the beta cells of the pancreas. Insulin plays a role in metabolic functions such as glucose uptake, glycolysis, glycogenesis, lipogenesis, and protein synthesis. Exogenous insulin may be needed for individuals with diabetes mellitus, in whom there is a deficiency in endogenous insulin or increased insulin resistance. Insulin precursor (proinsulin) C-peptideC-peptideThe middle segment of proinsulin that is between the n-terminal b-chain and the c-terminal a-chain. It is a pancreatic peptide of about 31 residues, depending on the species. Upon proteolytic cleavage of proinsulin, equimolar insulin and c-peptide are released. C-peptide immunoassay has been used to assess pancreatic beta cell function in diabetic patients with circulating insulin antibodies or exogenous insulin. Half-life of c-peptide is 30 min, almost 8 times that of insulin.Diabetes Mellitus is cleaved in GA).
Addition of a functional group
ProteinsProteinsLinear polypeptides that are synthesized on ribosomes and may be further modified, crosslinked, cleaved, or assembled into complex proteins with several subunits. The specific sequence of amino acids determines the shape the polypeptide will take, during protein folding, and the function of the protein.Energy Homeostasis are further modified by the covalent addition of functional groups and other molecules.
MethylationMethylationAddition of methyl groups. In histo-chemistry methylation is used to esterify carboxyl groups and remove sulfate groups by treating tissue sections with hot methanol in the presence of hydrochloric acid. .Glucocorticoids
Ubiquitylation
Glycosylation
Lipidation
Common sites that are modified:
Hydroxyl groups in serineSerineA non-essential amino acid occurring in natural form as the l-isomer. It is synthesized from glycine or threonine. It is involved in the biosynthesis of purines; pyrimidines; and other amino acids.Synthesis of Nonessential Amino Acids, threonine, and tyrosineTyrosineA non-essential amino acid. In animals it is synthesized from phenylalanine. It is also the precursor of epinephrine; thyroid hormones; and melanin.Synthesis of Nonessential Amino Acids
Amine groups in lysine, arginineArginineAn essential amino acid that is physiologically active in the l-form.Urea Cycle, and histidine
Carboxylate groups in aspartateAspartateOne of the non-essential amino acids commonly occurring in the l-form. It is found in animals and plants, especially in sugar cane and sugar beets. It may be a neurotransmitter.Synthesis of Nonessential Amino Acids and glutamateGlutamateDerivatives of glutamic acid. Included under this heading are a broad variety of acid forms, salts, esters, and amides that contain the 2-aminopentanedioic acid structure.Synthesis of Nonessential Amino Acids
The N- and C-terminals
General principles:
Hydrophobic groups may help a protein incorporate into a membrane.
Addition of cofactors can enhance enzymatic activity.
Phosphorylation
Addition of phosphoryl group (most common)
Common functions:
Regulates enzymatic activity
Cellular energy exchange: ATP, guanosine triphosphate (GTP), nicotinamide adenine dinucleotideNicotinamide adenine dinucleotideA coenzyme composed of ribosylnicotinamide 5′-diphosphate coupled to adenosine 5′-phosphate by pyrophosphate linkage. It is found widely in nature and is involved in numerous enzymatic reactions in which it serves as an electron carrier by being alternately oxidized (NAD+) and reduced (NADH).Pentose Phosphate PathwayphosphatePhosphateInorganic salts of phosphoric acid.Electrolytes (NADPHNADPHNicotinamide adenine dinucleotide phosphate. A coenzyme composed of ribosylnicotinamide 5′-phosphate (nmn) coupled by pyrophosphate linkage to the 5′-phosphate adenosine 2.Pentose Phosphate Pathway)
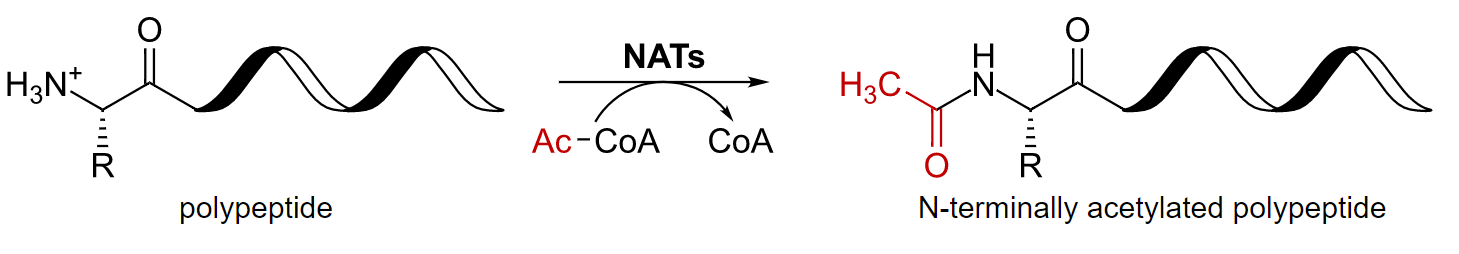
AcetylationAcetylationFormation of an acetyl derivative.Chloramphenicol and methylationMethylationAddition of methyl groups. In histo-chemistry methylation is used to esterify carboxyl groups and remove sulfate groups by treating tissue sections with hot methanol in the presence of hydrochloric acid. .Glucocorticoids
Addition of acetyl group or methyl group
Common functions:
Activates many pharmaceuticals
Regulates geneGeneA category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms.Basic Terms of Genetics expression and protein synthesisSynthesisPolymerase Chain Reaction (PCR)
Histone modifications
Acetylation of a polypeptide
CoA: coenzyme A NAT: N-terminal acetyltransferases
Image: “Protein-acetylation-nterminal” by Hbf878. License: CC0 1.0
Ubiquitylation
Addition of ubiquitin
Functions:
Targets proteinsProteinsLinear polypeptides that are synthesized on ribosomes and may be further modified, crosslinked, cleaved, or assembled into complex proteins with several subunits. The specific sequence of amino acids determines the shape the polypeptide will take, during protein folding, and the function of the protein.Energy Homeostasis for degradation in proteasome
Histone modifications
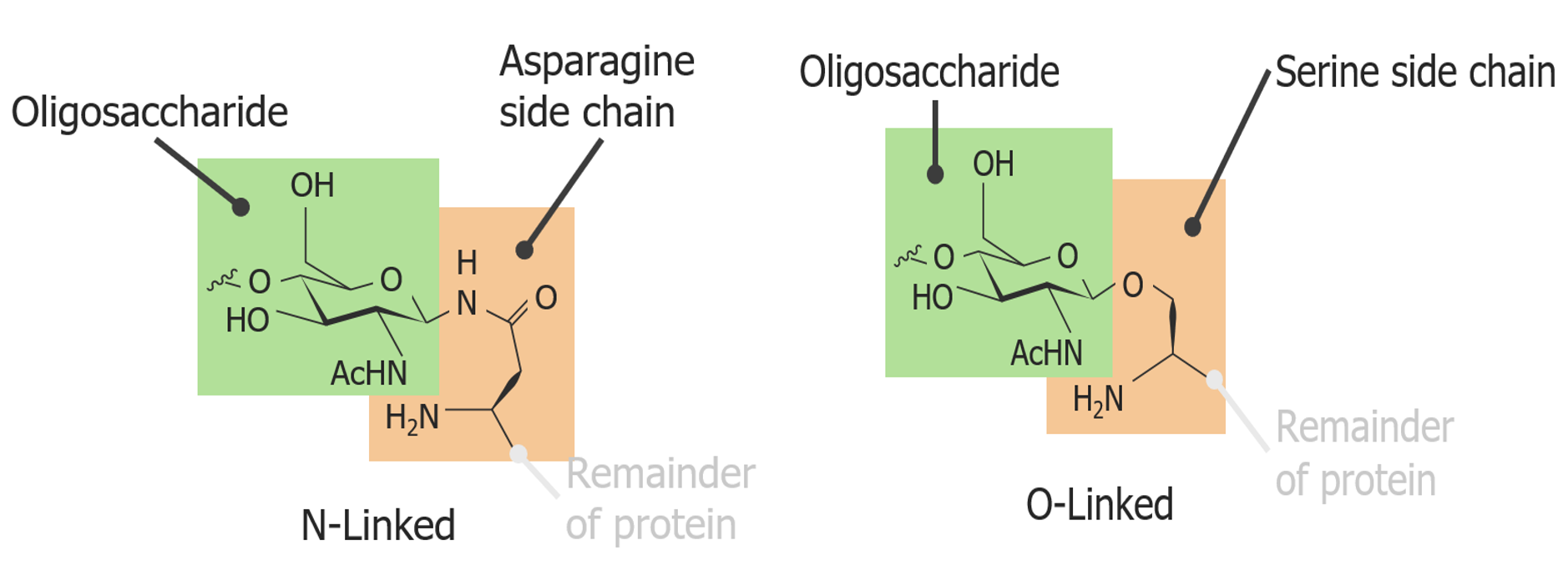
Glycosylation
Addition of carbohydrate to create a glycoprotein
Types of glycoproteinsGlycoproteinsConjugated protein-carbohydrate compounds including mucins, mucoid, and amyloid glycoproteins.Basics of Carbohydrates include:
N-glycoproteins: bound to nitrogenNitrogenAn element with the atomic symbol n, atomic number 7, and atomic weight [14. 00643; 14. 00728]. Nitrogen exists as a diatomic gas and makes up about 78% of the earth’s atmosphere by volume. It is a constituent of proteins and nucleic acids and found in all living cells.Urea Cycle on the side chain of asparagineAsparagineA non-essential amino acid that is involved in the metabolic control of cell functions in nerve and brain tissue. It is biosynthesized from aspartic acid and ammonia by asparagine synthetase.Synthesis of Nonessential Amino Acids
O-glycoproteins: bound to oxygen on the side chain of serineSerineA non-essential amino acid occurring in natural form as the l-isomer. It is synthesized from glycine or threonine. It is involved in the biosynthesis of purines; pyrimidines; and other amino acids.Synthesis of Nonessential Amino Acids or threonine
GlycosylatedGlycosylatedGlycopeptidesproteinsProteinsLinear polypeptides that are synthesized on ribosomes and may be further modified, crosslinked, cleaved, or assembled into complex proteins with several subunits. The specific sequence of amino acids determines the shape the polypeptide will take, during protein folding, and the function of the protein.Energy Homeostasis are commonly associated with the cell membraneCell MembraneA cell membrane (also known as the plasma membrane or plasmalemma) is a biological membrane that separates the cell contents from the outside environment. A cell membrane is composed of a phospholipid bilayer and proteins that function to protect cellular DNA and mediate the exchange of ions and molecules. The Cell: Cell Membrane or are secreted; common examples include:
HormonesHormonesHormones are messenger molecules that are synthesized in one part of the body and move through the bloodstream to exert specific regulatory effects on another part of the body. Hormones play critical roles in coordinating cellular activities throughout the body in response to the constant changes in both the internal and external environments. Hormones: Overview and Types
Immune systemImmune systemThe body’s defense mechanism against foreign organisms or substances and deviant native cells. It includes the humoral immune response and the cell-mediated response and consists of a complex of interrelated cellular, molecular, and genetic components.Primary Lymphatic Organs functions (e.g., found in some antibodiesAntibodiesImmunoglobulins (Igs), also known as antibodies, are glycoprotein molecules produced by plasma cells that act in immune responses by recognizing and binding particular antigens. The various Ig classes are IgG (the most abundant), IgM, IgE, IgD, and IgA, which differ in their biologic features, structure, target specificity, and distribution.Immunoglobulins: Types and Functions)
Cellular identity (e.g., ABO blood types)
Glycosylation affects many different cellular processes and is implicated in:
Cancer
DiabetesDiabetesDiabetes mellitus (DM) is a metabolic disease characterized by hyperglycemia and dysfunction of the regulation of glucose metabolism by insulin. Type 1 DM is diagnosed mostly in children and young adults as the result of autoimmune destruction of β cells in the pancreas and the resulting lack of insulin. Type 2 DM has a significant association with obesity and is characterized by insulin resistance.Diabetes Mellitus
Alzheimer’s disease
An N-linked versus an O-linked glycoprotein
Image by Lecturio.
Lipidation
Addition of a lipid molecule to create a proteolipidProteolipidProtein-lipid combinations abundant in brain tissue, but also present in a wide variety of animal and plant tissues. In contrast to lipoproteins, they are insoluble in water, but soluble in a chloroform-methanol mixture. The protein moiety has a high content of hydrophobic amino acids. The associated lipids consist of a mixture of glycerophosphates; cerebrosides; and sulfoglycosphingolipids; while lipoproteins contain phospholipids; cholesterol; and triglycerides.Orthopoxvirus
Typically occurs on proteinsProteinsLinear polypeptides that are synthesized on ribosomes and may be further modified, crosslinked, cleaved, or assembled into complex proteins with several subunits. The specific sequence of amino acids determines the shape the polypeptide will take, during protein folding, and the function of the protein.Energy Homeostasis associated with a phospholipid membrane
Examples of lipidation:
Addition of a glycosylphosphatidylinositol (GPI) “anchor”: commonly used to anchor cell surface proteinsProteinsLinear polypeptides that are synthesized on ribosomes and may be further modified, crosslinked, cleaved, or assembled into complex proteins with several subunits. The specific sequence of amino acids determines the shape the polypeptide will take, during protein folding, and the function of the protein.Energy Homeostasis
N-myristoylation: addition of a myristoyl group to some proteinsProteinsLinear polypeptides that are synthesized on ribosomes and may be further modified, crosslinked, cleaved, or assembled into complex proteins with several subunits. The specific sequence of amino acids determines the shape the polypeptide will take, during protein folding, and the function of the protein.Energy Homeostasis involved in signal transductionTransductionThe transfer of bacterial DNA by phages from an infected bacterium to another bacterium. This also refers to the transfer of genes into eukaryotic cells by viruses. This naturally occurring process is routinely employed as a gene transfer technique.Bacteriology, oncogenesisOncogenesisThe origin, production or development of cancer through genotypic and phenotypic changes which upset the normal balance between cell proliferation and cell death. Carcinogenesis generally requires a constellation of steps, which may occur quickly or over a period of many years.Carcinogenesis, and host defense
Prenylation or palmitoylation: addition of a prenyl or a palmitic acidPalmitic acidA common saturated fatty acid found in fats and waxes including olive oil, palm oil, and body lipids.Fatty Acids and Lipids group to membrane proteinsProteinsLinear polypeptides that are synthesized on ribosomes and may be further modified, crosslinked, cleaved, or assembled into complex proteins with several subunits. The specific sequence of amino acids determines the shape the polypeptide will take, during protein folding, and the function of the protein.Energy Homeostasis, making them more hydrophobic
Abnormalities in post-translational modification and/or protein foldingProtein foldingProcesses involved in the formation of tertiary protein structure.Proteins and Peptides or sorting can lead to a number of clinically important medical conditions.
Protease inhibitorsProtease InhibitorsCompounds which inhibit or antagonize biosynthesis or actions of proteases (endopeptidases).Anti-HIV Drugs:HIVHIVAnti-HIV Drugs uses the process of proteolysis during its life cycle to create functional structural proteinsStructural proteinsProteins and Peptides from precursors. These proteasesProteasesProteins and Peptides are a target of the anti-HIV drugsAnti-HIV drugsAntiretroviral therapy (ART) targets the replication cycle of the human immunodeficiency virus (HIV) and is classified based on the viral enzyme or mechanism that is inhibited. The goal of therapy is to suppress viral replication to reach the outcome of undetected viral load. Anti-HIV Drugs called protease inhibitorsProtease InhibitorsCompounds which inhibit or antagonize biosynthesis or actions of proteases (endopeptidases).Anti-HIV Drugs.
Alzheimer’s disease: neurodegenerative disease resulting in dementiaDementiaMajor neurocognitive disorders (NCD), also known as dementia, are a group of diseases characterized by decline in a person’s memory and executive function. These disorders are progressive and persistent diseases that are the leading cause of disability among elderly people worldwide.Major Neurocognitive Disorders: It is thought that misfolded and/or abnormally modified proteinsProteinsLinear polypeptides that are synthesized on ribosomes and may be further modified, crosslinked, cleaved, or assembled into complex proteins with several subunits. The specific sequence of amino acids determines the shape the polypeptide will take, during protein folding, and the function of the protein.Energy Homeostasis, including the β-amyloid peptide and tau proteinsProteinsLinear polypeptides that are synthesized on ribosomes and may be further modified, crosslinked, cleaved, or assembled into complex proteins with several subunits. The specific sequence of amino acids determines the shape the polypeptide will take, during protein folding, and the function of the protein.Energy Homeostasis, are associated with Alzheimer’s disease. Whether Alzheimer’s disease results in increased misfolded proteinsMisfolded ProteinsCell Injury and Death or the misfolded proteinsMisfolded ProteinsCell Injury and Death cause the disease is still being explored.
Parkinson’s disease: progressive neurodegenerative movement disorder that presents with tremors, stiffness, and slowing of movement: Parkinson’s disease is thought to be caused at least in part by accumulation of a protein called α-synuclein in the neuronsNeuronsThe basic cellular units of nervous tissue. Each neuron consists of a body, an axon, and dendrites. Their purpose is to receive, conduct, and transmit impulses in the nervous system.Nervous System: Histology of the nigrostriatal pathwayNigrostriatal pathwayBasal Ganglia: Anatomy, leading to the death of these neuronsNeuronsThe basic cellular units of nervous tissue. Each neuron consists of a body, an axon, and dendrites. Their purpose is to receive, conduct, and transmit impulses in the nervous system.Nervous System: Histology. Misfolding of α-synuclein leads to the formation of insoluble aggregates, which accumulate and disrupt signaling.
CysticCysticFibrocystic ChangefibrosisFibrosisAny pathological condition where fibrous connective tissue invades any organ, usually as a consequence of inflammation or other injury.Bronchiolitis Obliterans (CF):autosomal recessiveAutosomal recessiveAutosomal inheritance, both dominant and recessive, refers to the transmission of genes from the 22 autosomal chromosomes. Autosomal recessive diseases are only expressed when 2 copies of the recessive allele are inherited.Autosomal Recessive and Autosomal Dominant Inheritance disorder caused by mutations in the CFTRgeneGeneA category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms.Basic Terms of Genetics. The mutations lead to dysfunction of chlorideChlorideInorganic compounds derived from hydrochloric acid that contain the Cl- ion.ElectrolyteschannelsChannelsThe Cell: Cell Membrane, which results in hyperviscous mucus and the accumulation of secretions. There are 5 classes of mutations. Class II is a group of mutations that cause abnormal post-translational processing; because of these abnormalities, the proteinsProteinsLinear polypeptides that are synthesized on ribosomes and may be further modified, crosslinked, cleaved, or assembled into complex proteins with several subunits. The specific sequence of amino acids determines the shape the polypeptide will take, during protein folding, and the function of the protein.Energy Homeostasis are not brought to the correct cellular locations (and are often defective). This class includes the common mutationMutationGenetic mutations are errors in DNA that can cause protein misfolding and dysfunction. There are various types of mutations, including chromosomal, point, frameshift, and expansion mutations. Types of Mutations F508del. Common presentations of CF include chronic respiratory infectionsInfectionsInvasion of the host organism by microorganisms or their toxins or by parasites that can cause pathological conditions or diseases.Chronic Granulomatous Disease, failure to thriveFailure to ThriveFailure to thrive (FTT), or faltering growth, describes suboptimal weight gain and growth in children. The majority of cases are due to inadequate caloric intake; however, genetic, infectious, and oncological etiologies are also common. Failure to Thrive, and pancreatic insufficiency.
References
Alberts, B., Heald, R., Johnson, A., Morgan, D., Raff, M., Roberts, K., Walter, P., & Wilson, J. (2022). Molecular biology of the cell (7th ed.). W.W. Norton & Company.
Bruno, P. S., et al. (2025). Post-translational modifications of proteins orchestrate all hallmarks of cancer. Life, 15(1), 126. https://doi.org/10.3390/life15010126
Li, Y., et al. (2023). Advances in post-translational modifications of proteins and cancer immunotherapy. Frontiers in Immunology, 14. https://doi.org/10.3389/fimmu.2023.1229397
Peana, M., et al. (2024). Interplay of metal ions and posttranslational modifications in proteins. European Journal of Inorganic Chemistry, 27(31), e202400175. https://doi.org/10.1002/ejic.202400175
Tahiri, E., et al. (2025). Regulation of synaptic NMDA receptor activity by post-translational modifications. Neurochemical Research, 50(2), 110. https://doi.org/10.1007/s11064-025-04346-6
Zafar, S., et al. (2024). Current technologies unraveling the significance of post-translational modifications (PTMs) as crucial players in neurodegeneration. Biomolecules, 14(1), 118. https://doi.org/10.3390/biom14010118
Create your free account or log in to continue reading!