The hemophilias are a group of inherited, or sometimes acquired, disorders of secondary hemostasis Secondary hemostasis The coagulation cascade is a series of reactions that ultimately generates a strong, cross-linked fibrin clot. Hemostasis due to deficiency of specific clotting factors. Hemophilia A is a deficiency of factor VIII Factor VIII Factor VIII of blood coagulation. Antihemophilic factor that is part of the factor viii/von Willebrand factor complex. Factor VIII is produced in the liver and acts in the intrinsic pathway of blood coagulation. It serves as a cofactor in factor X activation and this action is markedly enhanced by small amounts of thrombin. Hemostasis, hemophilia B a deficiency of factor IX Factor IX Storage-stable blood coagulation factor acting in the intrinsic pathway of blood coagulation. Its activated form, ixa, forms a complex with factor VIII and calcium on platelet factor 3 to activate factor X to Xa. Hemostasis, and hemophilia C a deficiency of factor XI. Patients Patients Individuals participating in the health care system for the purpose of receiving therapeutic, diagnostic, or preventive procedures. Clinician–Patient Relationship present with bleeding events that may be spontaneous or associated with minor or major trauma. Management is focused mainly on treatment of acute bleeding events and prevention of bleeding events via replacement of deficient factors.
Last updated: Dec 15, 2025
Hemophilias are a group of disorders of secondary hemostasis Secondary hemostasis The coagulation cascade is a series of reactions that ultimately generates a strong, cross-linked fibrin clot. Hemostasis due to the deficiency of specific clotting factors.
About 400 children per year are born with hemophilia in the United States.
3 different types have been described:
Genetic mutation Mutation Genetic mutations are errors in DNA that can cause protein misfolding and dysfunction. There are various types of mutations, including chromosomal, point, frameshift, and expansion mutations. Types of Mutations resulting in deficiency of clotting factors:
Acquired hemophilia:
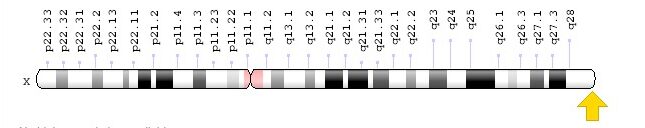
Location of F8 gene for coagulation factor VIII: Xq28, resulting in hemophilia A
Image: “F8 gene location” by Genome Decoration Page/NCBI. License: Public Domain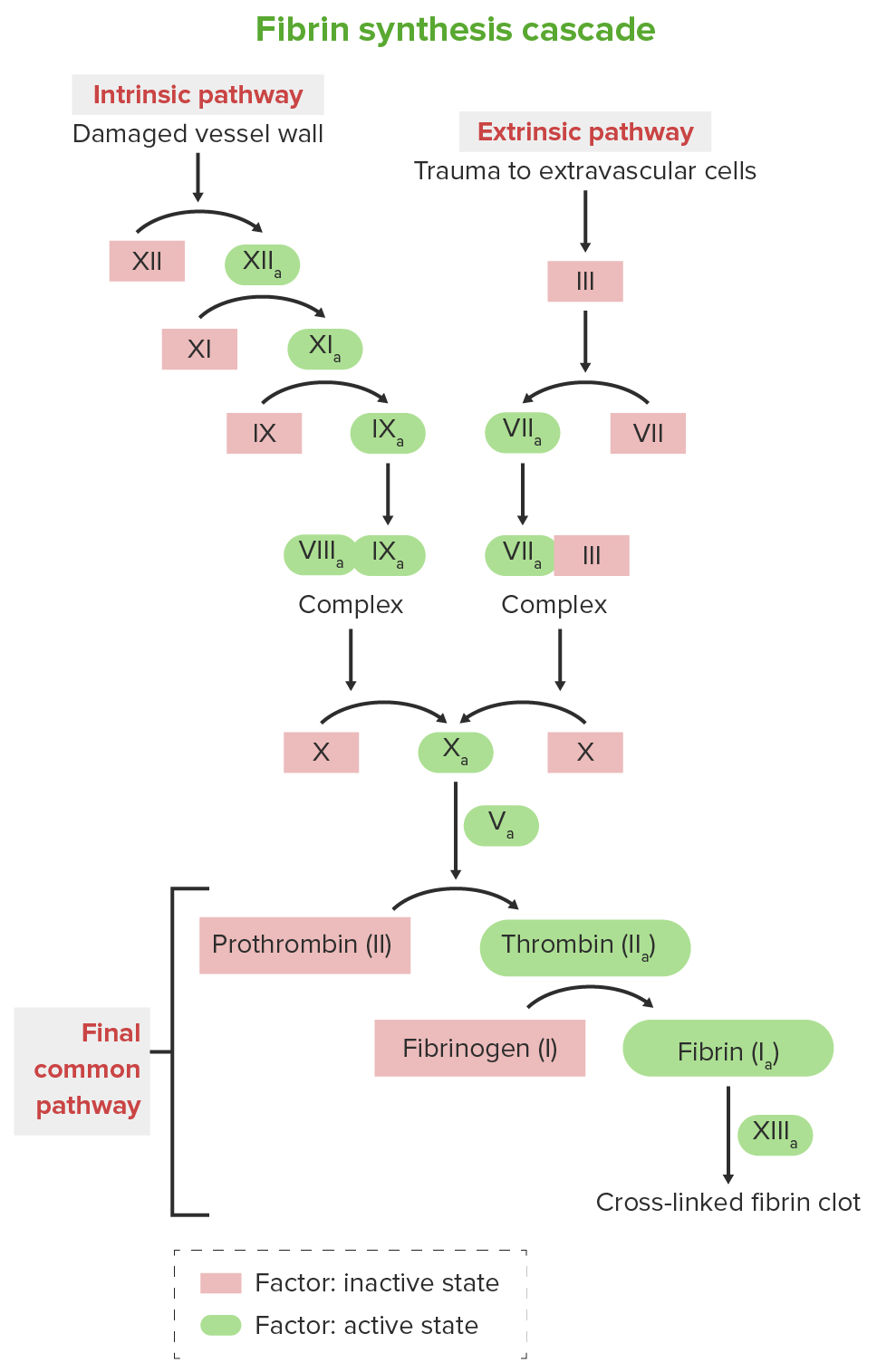
Clotting cascade
Image by Lecturio.Presentation is based mainly on the severity of the factor deficiency.
Mild to moderate deficiency:
More severe deficiency:

Extensive subcutaneous hematoma/ecchymosis of the limbs, thorax, and abdomen in a patient with hemophilia
Image: “Extensive subcutaneous ecchymoses” by Arthur Bloom Hemophilia Centre, School of Medicine, Cardiff University, University Hospital of Wales, Cardiff, CF14 4XN, UK. License: CC BY 2.0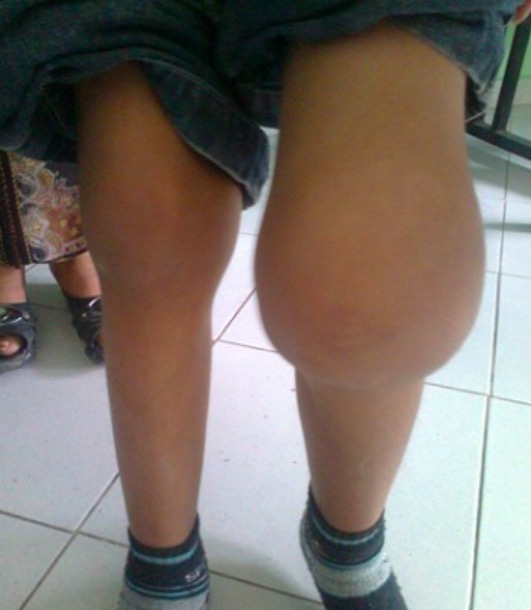
Hemarthrosis in a 6-year-old boy
Image: “ Hémarthrose” by Service de Pédiatrie, Hôpital Al Farabi, CHU Mohamed VI, Université Mohamed I, Oujda, Maroc. License: CC BY 2.0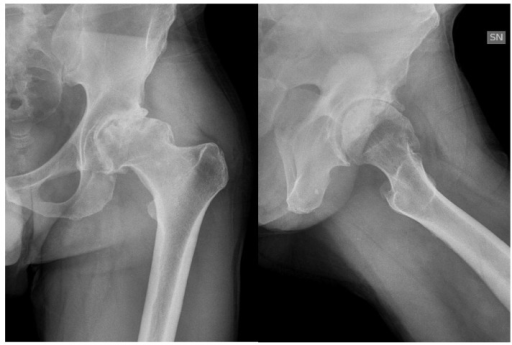
Hip arthropathy in hemophilia: symptomatic arthropathy of the left hip secondary to femoroacetabular impingement in a 46-year-old individual affected by severe hemophilia A with inhibitors
Image: “jcm-06-00044-f001” by Christian Carulli, Anna Rosa Rizzo, Massimo Innocenti. License: CC BY 4.0The diagnosis of hemophilia is made on the basis of clinical suspicion (usually due to a bleeding event) followed by demonstration of deficient factor levels (< 40%).