


As hemofilias são um grupo de doenças hereditárias, ou às vezes adquiridas, da hemostase secundária devido à deficiência de fatores de coagulação específicos. A hemofilia A é a deficiência do fator VIII, a hemofilia B a deficiência do fator IX e a hemofilia C a deficiência do fator XI. Os pacientes apresentam eventos hemorrágicos que podem ser espontâneos ou associados a traumas minor ou major. A abordagem terapêutica foca-se principalmente no tratamento de eventos hemorrágicos agudos e na prevenção de eventos hemorrágicos através da reposição de fatores em défice.
Last updated: Dec 15, 2025
As hemofilias são um grupo de doenças da hemostase secundária devido à deficiência de fatores de coagulação específicos.
Nos Estados Unidos nascem cerca de 400 crianças por ano com hemofilia.
Foram descritos 3 tipos diferentes:
Mutação genética que resulta em deficiência de fatores de coagulação:
Hemofilia adquirida:
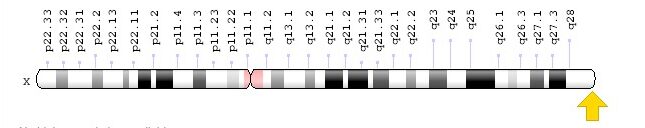
Localização do gene F8 para o fator de coagulação VIII: Xq28, resultando em hemofilia A
Imagem: “F8 gene location” por Genome Decoration Page/NCBI. Licença: Domínio Público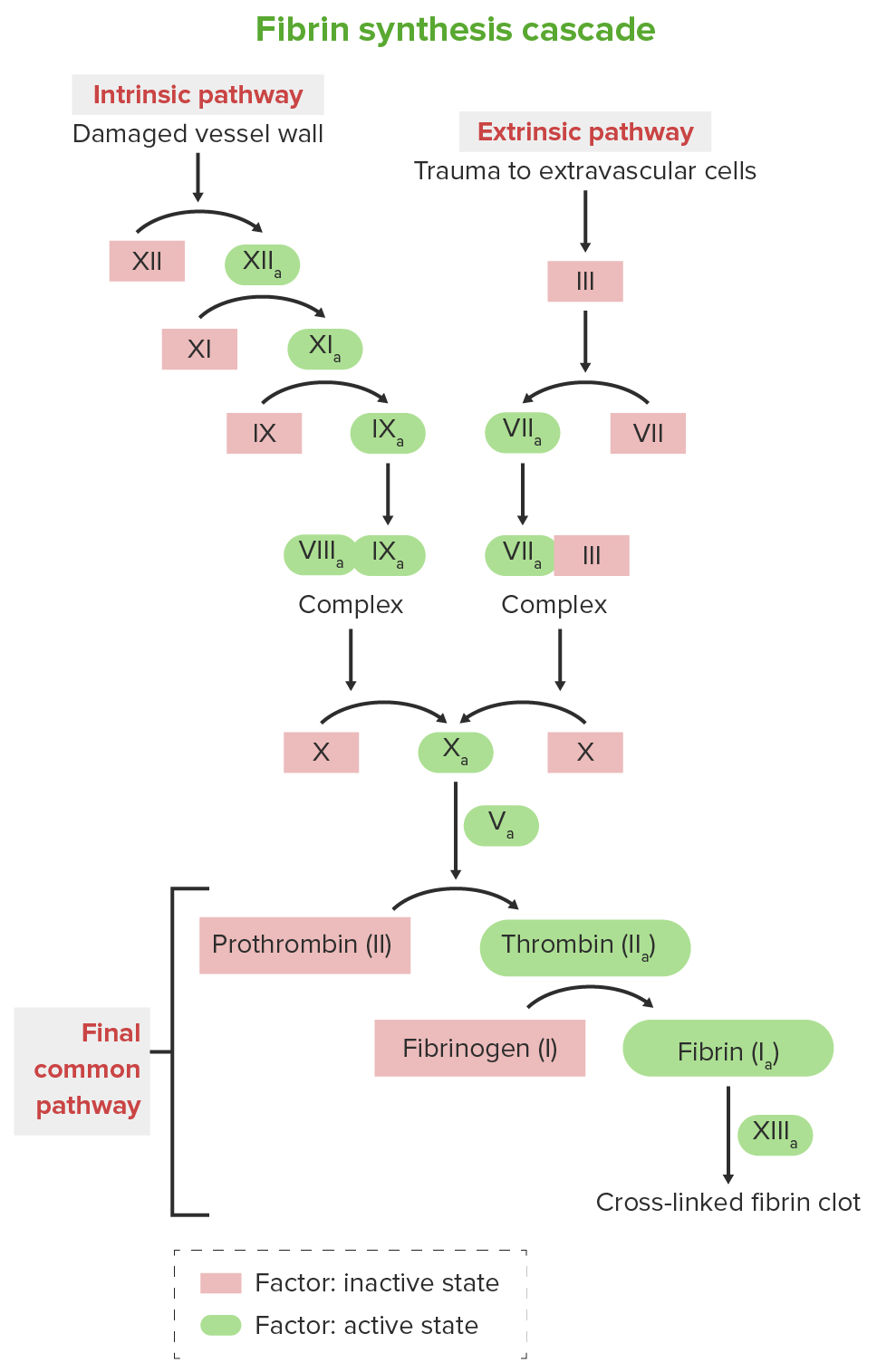
Cascata de coagulação
Imagem por Lecturio.A apresentação depende principalmente na gravidade da deficiência do fator.
Deficiência ligeira a moderada:
Deficiência mais MAIS Androgen Insensitivity Syndrome grave:

Hematoma subcutâneo extenso/equimose dos membros, tórax e abdómen em paciente com hemofilia
Imagem : “Extensive subcutaneous ecchymoses” por Arthur Bloom Hemophilia Centre, School of Medicine, Cardiff University, University Hospital of Wales, Cardiff, CF14 4XN, UK. Licença: CC BY 2.0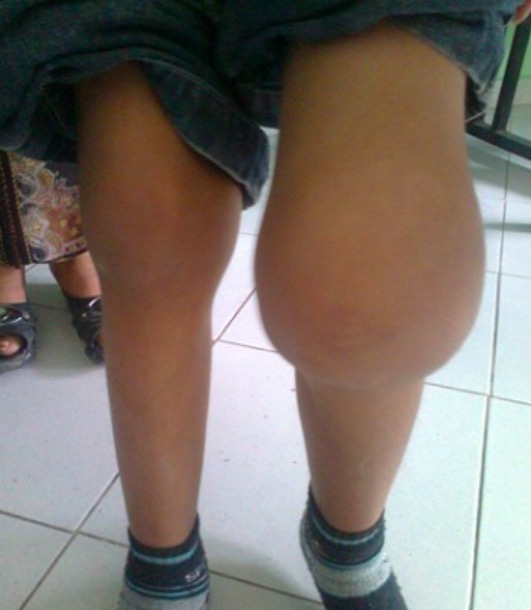
Hemartrose em menino de 6 anos
Imagem : “Hémartrose” por Service de Pédiatrie, Hôpital Al Farabi, CHU Mohamed VI, Université Mohamed I, Oujda, Marrocos. Licença: CC BY 2.0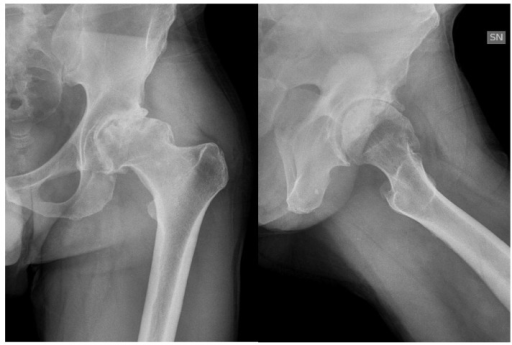
Artropatia da anca na hemofilia: artropatia sintomática da anca esquerda secundária a conflito fémuro-acetabular num indivíduo de 46 anos com hemofilia A grave com inibidores
Imagem: “jcm-06-00044-f001” por Christian Carulli, Anna Rosa Rizzo, Massimo Innocenti. Licença: CC BY 4.0O diagnóstico de hemofilia é feito com base na suspeita clínica (geralmente devido a um evento hemorrágico) seguida pela demonstração de níveis deficitários de fatores (< 40%).
