Las hemofilias son un grupo de trastornos hereditarios, o a veces adquiridos, de la hemostasia secundaria debido a deficiencia de factores de coagulación específicos. La hemofilia A es una deficiencia del factor VIII Factor VIII Factor VIII of blood coagulation. Antihemophilic factor that is part of the factor viii/von Willebrand factor complex. Factor VIII is produced in the liver and acts in the intrinsic pathway of blood coagulation. It serves as a cofactor in factor X activation and this action is markedly enhanced by small amounts of thrombin. Hemostasis, la hemofilia B una deficiencia del factor IX Factor IX Storage-stable blood coagulation factor acting in the intrinsic pathway of blood coagulation. Its activated form, ixa, forms a complex with factor VIII and calcium on platelet factor 3 to activate factor X to Xa. Hemostasis, y la hemofilia C una deficiencia del factor XI. Los LOS Neisseria pacientes presentan eventos hemorrágicos que pueden ser espontáneos o estar asociados a un traumatismo menor o mayor. El tratamiento se centra principalmente en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum tratar los LOS Neisseria eventos hemorrágicos agudos y en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum la prevención de los LOS Neisseria eventos hemorrágicos mediante la sustitución de los LOS Neisseria factores deficientes.
Last updated: Dec 15, 2025
Las hemofilias son un grupo de trastornos de la hemostasia secundaria debidos a la deficiencia de factores de coagulación específicos.
En EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum Estados Unidos nacen unos 400 niños al AL Amyloidosis año con hemofilia.
Se han descrito 3 tipos diferentes:
Mutación genética que resulta en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum una deficiencia de factores de coagulación:
Hemofilia adquirida:
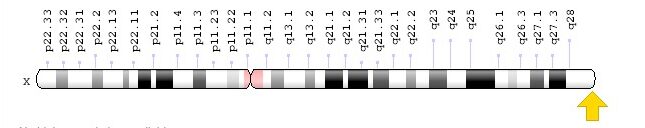
Localización del gen F8 para el factor VIII de coagulación: Xq28, que da lugar a la hemofilia A
Imagen: “F8 gene location” por Genome Decoration Page/NCBI. Licencia: Dominio Público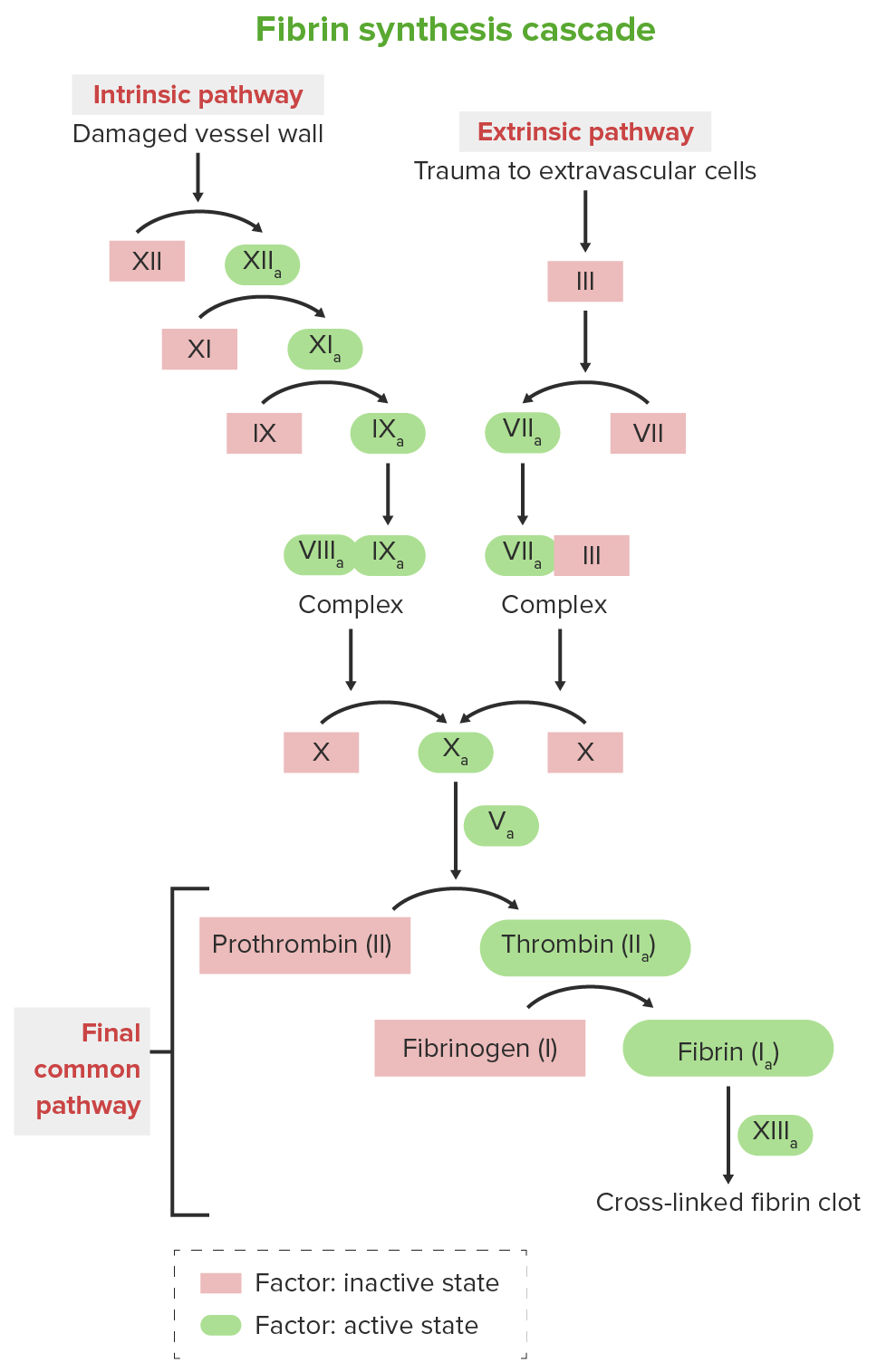
Cascada de la coagulación
Imagen por Lecturio.La presentación se basa principalmente en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum la gravedad de la deficiencia del factor.
Deficiencia leve a moderada:
Deficiencia más severa:
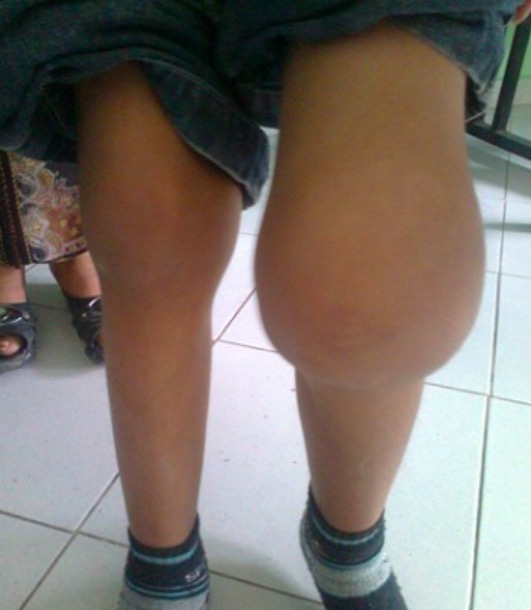
Hemartrosis en un niño de 6 años
Imagen: “ Hémarthrose” por Service de Pédiatrie, Hôpital Al Farabi, CHU Mohamed VI, Université Mohamed I, Oujda, Maroc. Licencia: CC BY 2.0
Hematoma/equimosis subcutánea extensa de las extremidades, el tórax y el abdomen en un paciente con hemofilia
Imagen: “Extensive subcutaneous ecchymoses” por Arthur Bloom Hemophilia Centre, School of Medicine, Cardiff University, University Hospital of Wales, Cardiff, CF14 4XN, UK. Licencia: CC BY 2.0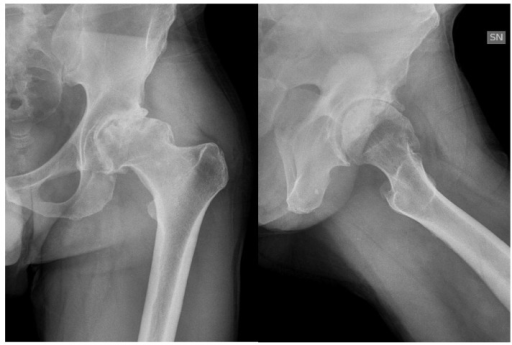
Artropatía de la cadera en la hemofilia: artropatía sintomática de la cadera izquierda secundaria a un pinzamiento femoroacetabular en un individuo de 46 años afectado por hemofilia A grave con inhibidores
Imagen: “jcm-06-00044-f001” por Christian Carulli, Anna Rosa Rizzo, Massimo Innocenti. Licencia: CC BY 4.0El diagnóstico de la hemofilia se hace HACE Altitude Sickness sobre la base de la sospecha clínica (generalmente debido a un evento hemorrágico) seguida de la demostración de niveles deficientes de un factor (< 40%).