Thrombocytopenia occurs when the platelet count is < 150,000 per microliter. The normal range for platelets Platelets Platelets are small cell fragments involved in hemostasis. Thrombopoiesis takes place primarily in the bone marrow through a series of cell differentiation and is influenced by several cytokines. Platelets are formed after fragmentation of the megakaryocyte cytoplasm. Platelets: Histology is usually 150,000–450,000/µL of whole blood. Thrombocytopenia can be a result of decreased production, increased destruction, or splenic sequestration Splenic sequestration Severe Congenital Neutropenia of platelets Platelets Platelets are small cell fragments involved in hemostasis. Thrombopoiesis takes place primarily in the bone marrow through a series of cell differentiation and is influenced by several cytokines. Platelets are formed after fragmentation of the megakaryocyte cytoplasm. Platelets: Histology. Patients Patients Individuals participating in the health care system for the purpose of receiving therapeutic, diagnostic, or preventive procedures. Clinician–Patient Relationship are often asymptomatic until platelet counts Platelet counts The number of platelets per unit volume in a sample of venous blood. Coagulation Studies are < 50,000/µL. Clinical manifestations of thrombocytopenia include easy bruising Easy bruising Chédiak-Higashi Syndrome or bleeding, petechiae Petechiae Primary Skin Lesions, purpura, and when severe, spontaneous mucosal and/or internal bleeding. Diagnosis is made with a CBC and blood smear Blood smear Myeloperoxidase Deficiency; additional testing may be required to determine the underlying etiology if it is not evident from the clinical scenario. Management involves treating the underlying etiology and platelet transfusions.
Last updated: Dec 15, 2025
Thrombocytopenia is a deficiency of platelets Platelets Platelets are small cell fragments involved in hemostasis. Thrombopoiesis takes place primarily in the bone marrow through a series of cell differentiation and is influenced by several cytokines. Platelets are formed after fragmentation of the megakaryocyte cytoplasm. Platelets: Histology, typically defined as < 150,000 platelets Platelets Platelets are small cell fragments involved in hemostasis. Thrombopoiesis takes place primarily in the bone marrow through a series of cell differentiation and is influenced by several cytokines. Platelets are formed after fragmentation of the megakaryocyte cytoplasm. Platelets: Histology per microliter of whole blood.
Thrombocytopenia can be classified as mild, moderate, or severe on the basis of platelet counts Platelet counts The number of platelets per unit volume in a sample of venous blood. Coagulation Studies:
Thrombocytopenia can be caused by decreased production, increased destruction, or sequestration of platelets Platelets Platelets are small cell fragments involved in hemostasis. Thrombopoiesis takes place primarily in the bone marrow through a series of cell differentiation and is influenced by several cytokines. Platelets are formed after fragmentation of the megakaryocyte cytoplasm. Platelets: Histology.
| Mechanism | Examples |
|---|---|
| Decreased production | Failure of
bone marrow
Bone marrow
The soft tissue filling the cavities of bones. Bone marrow exists in two types, yellow and red. Yellow marrow is found in the large cavities of large bones and consists mostly of fat cells and a few primitive blood cells. Red marrow is a hematopoietic tissue and is the site of production of erythrocytes and granular leukocytes. Bone marrow is made up of a framework of connective tissue containing branching fibers with the frame being filled with marrow cells.
Bone Marrow: Composition and Hematopoiesis:
|
Decreased
thrombopoietin
Thrombopoietin
A humoral factor that stimulates the production of thrombocytes (blood platelets). Thrombopoietin stimulates the proliferation of bone marrow megakaryocytes and their release of blood platelets. The process is called thrombopoiesis.
Platelets: Histology:
|
|
Nutritional:
|
|
Congenital
Congenital
Chorioretinitis:
|
|
| Increased destruction | Immune-mediated:
|
| Consumption in thrombi |
|
| Sequestration in the spleen Spleen The spleen is the largest lymphoid organ in the body, located in the LUQ of the abdomen, superior to the left kidney and posterior to the stomach at the level of the 9th-11th ribs just below the diaphragm. The spleen is highly vascular and acts as an important blood filter, cleansing the blood of pathogens and damaged erythrocytes. Spleen: Anatomy |
|
| Mixed or unclear mechanisms | Infection-induced thrombocytopenia:
|
| Dilution |
|
| Pseudothrombocytopenia (a falsely low platelet count due to clumping) |
|
Under normal physiologic conditions, platelets Platelets Platelets are small cell fragments involved in hemostasis. Thrombopoiesis takes place primarily in the bone marrow through a series of cell differentiation and is influenced by several cytokines. Platelets are formed after fragmentation of the megakaryocyte cytoplasm. Platelets: Histology are produced from the fragmentation Fragmentation Chronic Apophyseal Injury of megakaryocytes in the bone marrow Bone marrow The soft tissue filling the cavities of bones. Bone marrow exists in two types, yellow and red. Yellow marrow is found in the large cavities of large bones and consists mostly of fat cells and a few primitive blood cells. Red marrow is a hematopoietic tissue and is the site of production of erythrocytes and granular leukocytes. Bone marrow is made up of a framework of connective tissue containing branching fibers with the frame being filled with marrow cells. Bone Marrow: Composition and Hematopoiesis.
Platelets Platelets Platelets are small cell fragments involved in hemostasis. Thrombopoiesis takes place primarily in the bone marrow through a series of cell differentiation and is influenced by several cytokines. Platelets are formed after fragmentation of the megakaryocyte cytoplasm. Platelets: Histology are the principle cells involved in formation of the initial platelet plug Platelet plug Hemostasis (i.e., primary hemostasis Primary hemostasis Hemostasis).
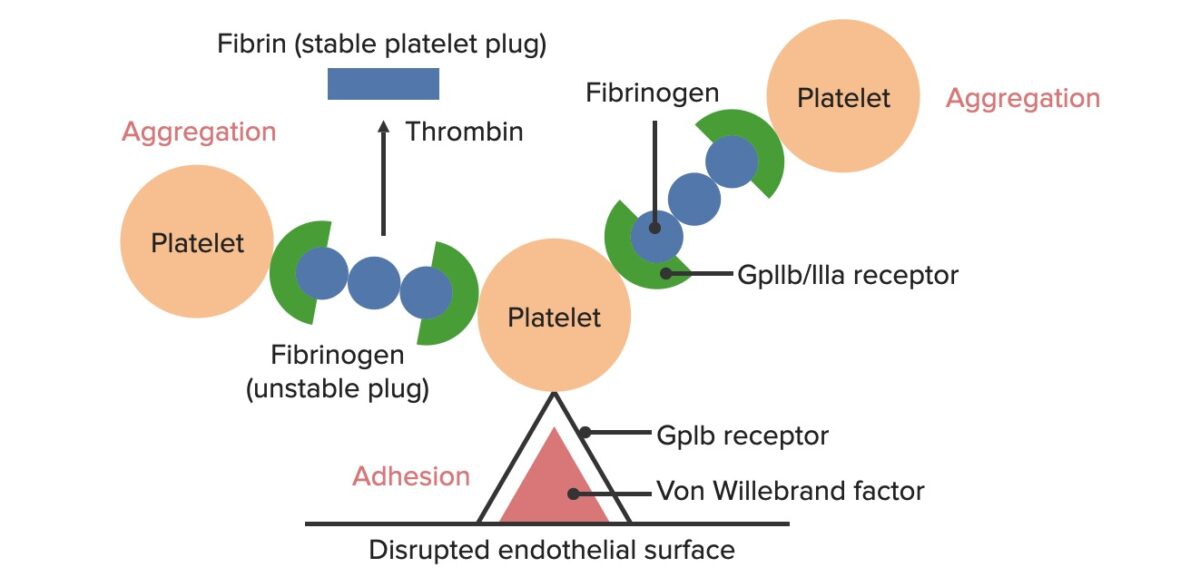
Formation of the temporary hemostatic plug:
The disrupted endothelial surface exposes VWF to the passing blood. Platelets bind to the VWF via their GpIb receptors and are activated. Platelet activation triggers them to secrete ADP, which stimulates the expression of the GpIIb/IIIa receptors on the platelets. The GpIIb/IIIa receptors bind to fibrinogen, which is able to bind a platelet on each end, causing platelets to aggregate. As more platelets are bound to one another, the platelet plug is generated. As the coagulation cascade is activated, thrombin converts the weaker fibrinogen into the stronger fibrin, creating a much more stable clot.
In thrombocytopenia, a platelet deficiency results in:
Drug-induced thrombocytopenia may be due to direct toxic effects on the bone marrow Bone marrow The soft tissue filling the cavities of bones. Bone marrow exists in two types, yellow and red. Yellow marrow is found in the large cavities of large bones and consists mostly of fat cells and a few primitive blood cells. Red marrow is a hematopoietic tissue and is the site of production of erythrocytes and granular leukocytes. Bone marrow is made up of a framework of connective tissue containing branching fibers with the frame being filled with marrow cells. Bone Marrow: Composition and Hematopoiesis (e.g., chemotherapy Chemotherapy Osteosarcoma) or to drug-dependent antibodies Antibodies Immunoglobulins (Igs), also known as antibodies, are glycoprotein molecules produced by plasma cells that act in immune responses by recognizing and binding particular antigens. The various Ig classes are IgG (the most abundant), IgM, IgE, IgD, and IgA, which differ in their biologic features, structure, target specificity, and distribution. Immunoglobulins: Types and Functions. Drug-dependent antibodies Antibodies Immunoglobulins (Igs), also known as antibodies, are glycoprotein molecules produced by plasma cells that act in immune responses by recognizing and binding particular antigens. The various Ig classes are IgG (the most abundant), IgM, IgE, IgD, and IgA, which differ in their biologic features, structure, target specificity, and distribution. Immunoglobulins: Types and Functions:
Unlike most drug-induced thrombocytopenias, HIT is associated with a markedly increased risk of thrombosis Thrombosis Formation and development of a thrombus or blood clot in the blood vessel. Epidemic Typhus rather than bleeding.
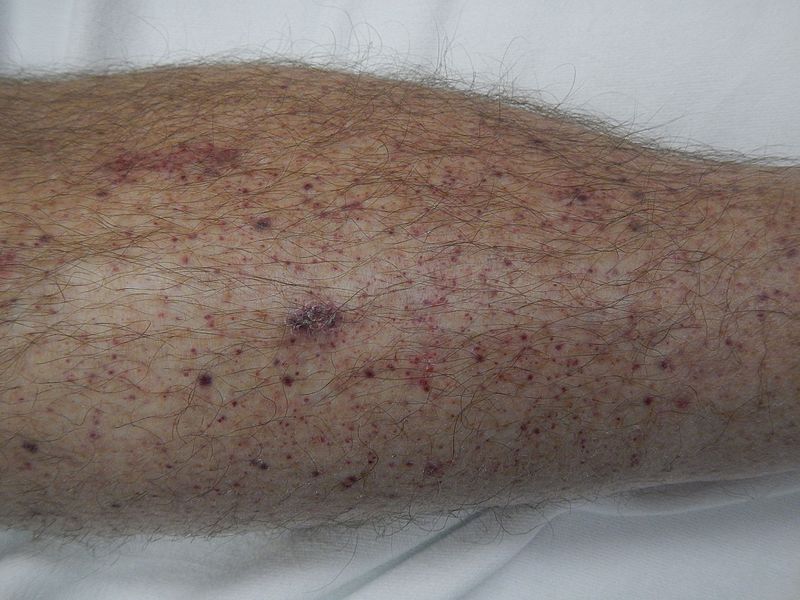
Petechiae on the lower leg of a patient with immune thrombocytopenic purpura
Image: “Petechiae on lower leg” by James Heilman, MD. License: CC BY-SA 4.0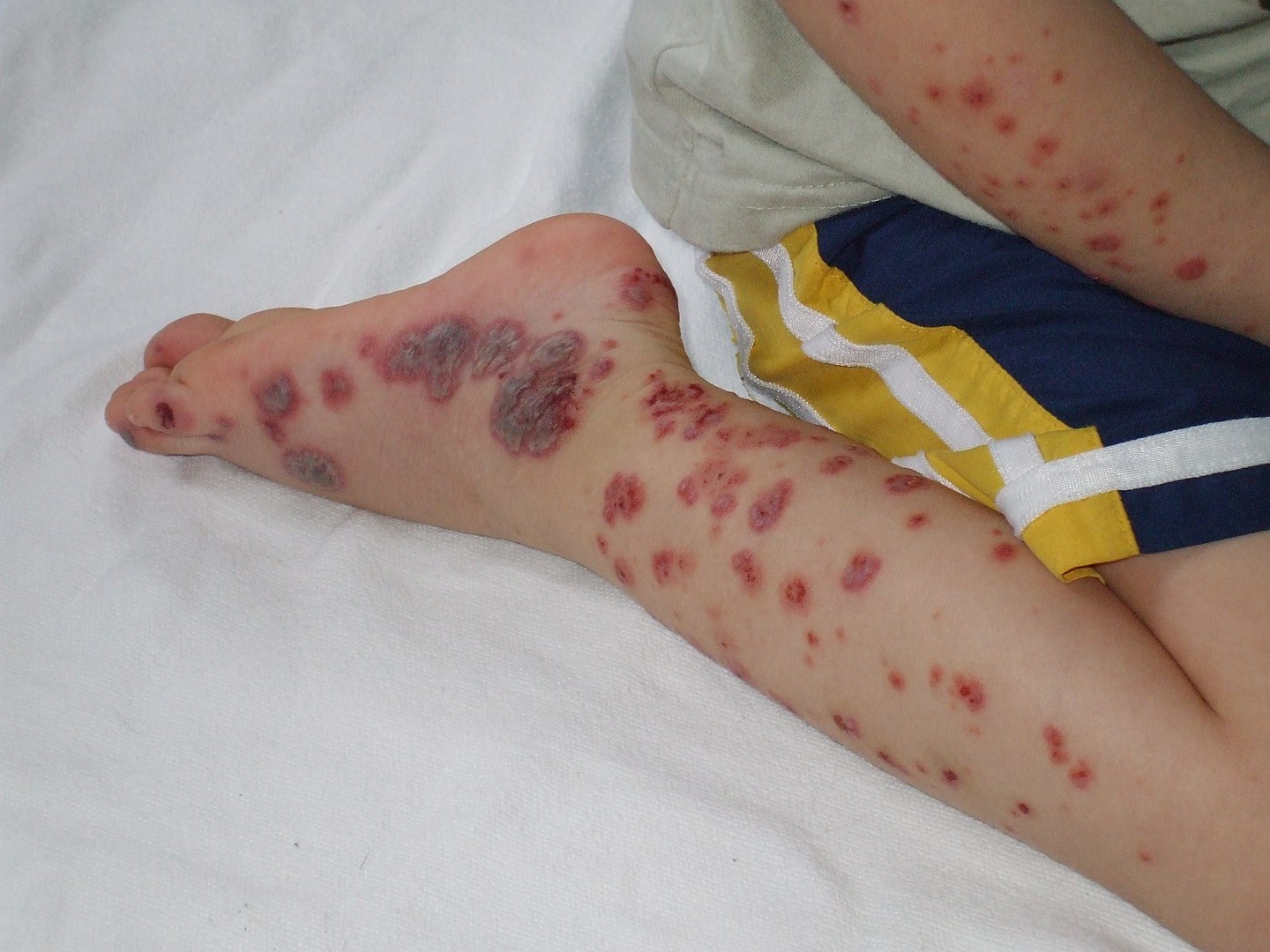
Purpura evident on the distal extremities of a pediatric patient
Image: “Purpura2” by Okwikikim. License: Public DomainThe diagnosis of thrombocytopenia itself is made with laboratory evaluations. Clues to the underlying etiology can be obtained from the history and exam, and additional lab testing can further aid in diagnosis.
The following tests should be ordered for all patients Patients Individuals participating in the health care system for the purpose of receiving therapeutic, diagnostic, or preventive procedures. Clinician–Patient Relationship with thrombocytopenia:
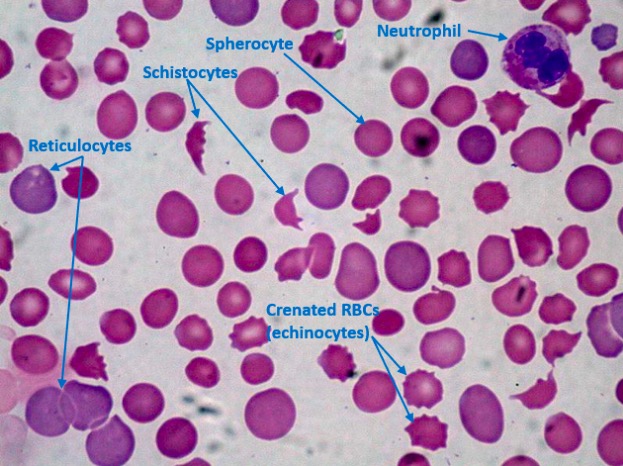
Peripheral blood film in a patient with DIC demonstrating thrombocytopenia, schistocytes, and spherocytes:
Many reticulocytes (immature RBCs) are present, indicating a brisk hematopoietic bone marrow response. Platelet counts are reduced (none seen here) because of consumption due to DIC. Normally, there should be at least 8 platelets in a single 1000×-oil-immersion field; 1 platelet indicates approximately 20,000 platelets/µL in the peripheral blood.
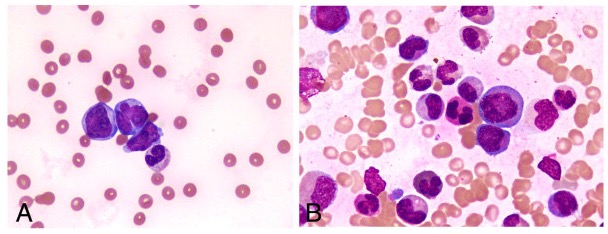
Peripheral smears showing a paucity of platelets
Image: “Peripheral blood smear and bone marrow aspirate” by Shabneez Hussain. License: CC BY 2.0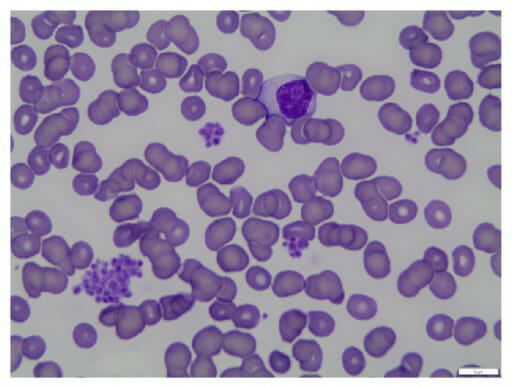
Blood smear of pseudothrombocytopenia: Pseudothrombocytopenia is caused by platelets clumping in vitro due to agglutination.
Image: “fig1: Peripheral blood smear (100x oil)” by Geok Chin Tan. License: CC BY 4.0The following tests should be ordered only as clinically indicated to help identify the underlying etiology.
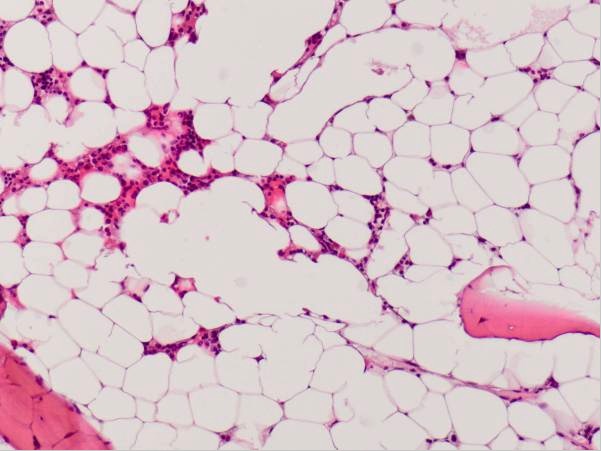
Hypocellular bone marrow in a patient with aplastic anemia
Image: “Hypocellular bone marrow showing only little hematopoiesis” by Department of Obstetrics of Gynaecology, Erasmus University Medical Center, PO Box 2040, 3000 CA Rotterdam, The Netherlands. License: CC BY 2.0Management of thrombocytopenia depends mainly on addressing the underlying cause, monitoring platelet counts Platelet counts The number of platelets per unit volume in a sample of venous blood. Coagulation Studies, and preventing/controlling any bleeding complications.