


A trombocitopenia ocorre quando a contagem de plaquetas é < 150.000 por microlitro. O intervalo normal para plaquetas é geralmente 150.000–450.000/µL de sangue total. A trombocitopenia pode resultar da diminuição da produção, do aumento da destruição ou do sequestro esplénico de plaquetas. Os pacientes geralmente são assintomáticos até que a contagem de plaquetas seja < 50.000/µL. As manifestações clínicas da trombocitopenia incluem hematomas ou hemorragia fáceis, petéquias, púrpura e, quando grave, hemorragia espontânea das mucosas e/ou interna. O diagnóstico é feito com um hemograma completo e com esfregaço de sangue; podem ser necessários exames adicionais para determinar a etiologia subjacente se não for evidente no cenário clínico. A abordagem terapêutica envolve o tratamento da etiologia subjacente e transfusões de plaquetas.
Last updated: Dec 15, 2025
A trombocitopenia é uma deficiência de plaquetas, tipicamente definida como < 150.000 plaquetas por microlitro de sangue total.
A trombocitopenia pode ser classificada como ligeira, moderada ou grave com base na contagem de plaquetas:
A trombocitopenia pode ser causada por diminuição da produção, aumento da destruição ou sequestro de plaquetas.
| Mecanismo | Exemplos |
|---|---|
| Diminuição da produção | Insuficiência da medula óssea:
|
Diminuição da trombopoietina:
|
|
Nutricional:
|
|
Congénita:
|
|
| Aumento da destruição | Imunomediado:
|
| Consumo por trombos |
|
| Sequestro esplénico |
|
| Mecanismos mistos ou pouco claros | Trombocitopenia induzida por infeção:
|
| Diluição |
|
| Pseudotrombocitopenia (uma contagem de plaquetas falsamente baixa devido a aglomeração) |
|
Em condições fisiológicas normais, as plaquetas são produzidas a partir da fragmentação de megacariócitos na medula óssea.
As plaquetas são as principais células envolvidas na formação do tampão plaquetário inicial (isto é, hemostase primária).
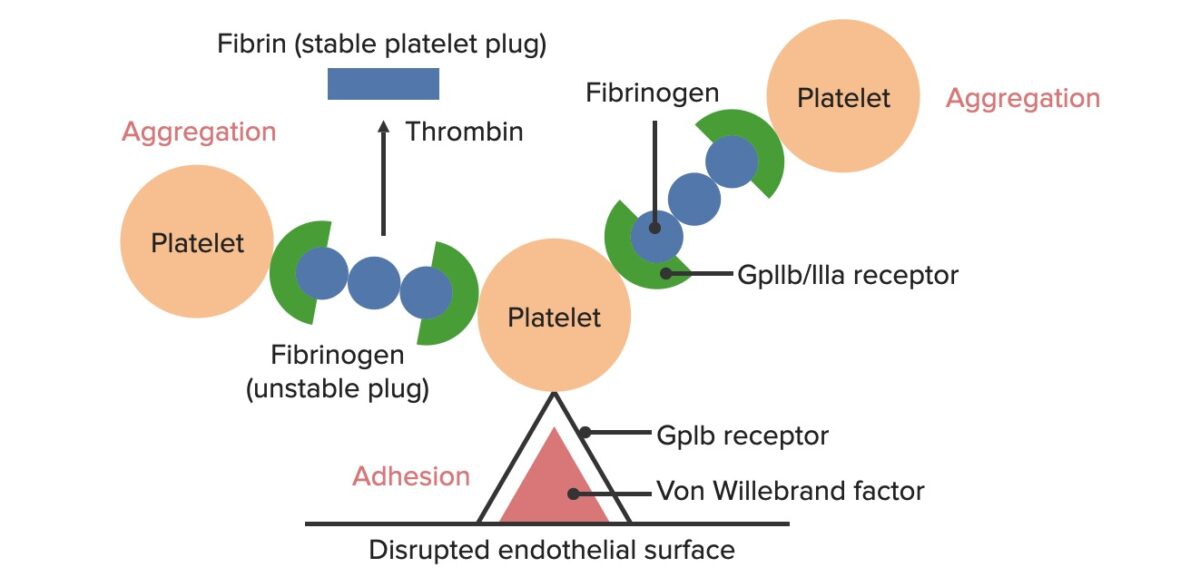
Formação do tampão hemostático temporário:
A superfície endotelial afetada expõe o FvW ao sangue que passa. As plaquetas ligam-se ao FvW através dos seus recetores GpIb e são ativadas. A ativação plaquetária desencadeia a secreção de ADP, que estimula a expressão dos recetores GpIIb/IIIa nas plaquetas. Os recetores GpIIb/IIIa ligam-se ao fibrinogénio, que é capaz de ligar uma plaqueta em cada extremidade, fazendo com que as plaquetas se agreguem. À medida que mais plaquetas são ligadas umas às outras, gera-se o tampão plaquetário. À medida que a cascata de coagulação é ativada, a trombina converte o fibrinogénio mais fraco na fibrina mais forte, criando um coágulo muito mais estável.
Na trombocitopenia, uma deficiência de plaquetas resulta em:
A trombocitopenia induzida por fármacos pode dever-se a efeitos tóxicos diretos na medula óssea (por exemplo, quimioterapia) ou a anticorpos dependentes de fármacos. Os anticorpos dependentes de fármacos:
Ao contrário da maioria das trombocitopenias induzidas por fármacos, a TIH está associada a um risco marcadamente aumentado de trombose em vez de hemorragia.
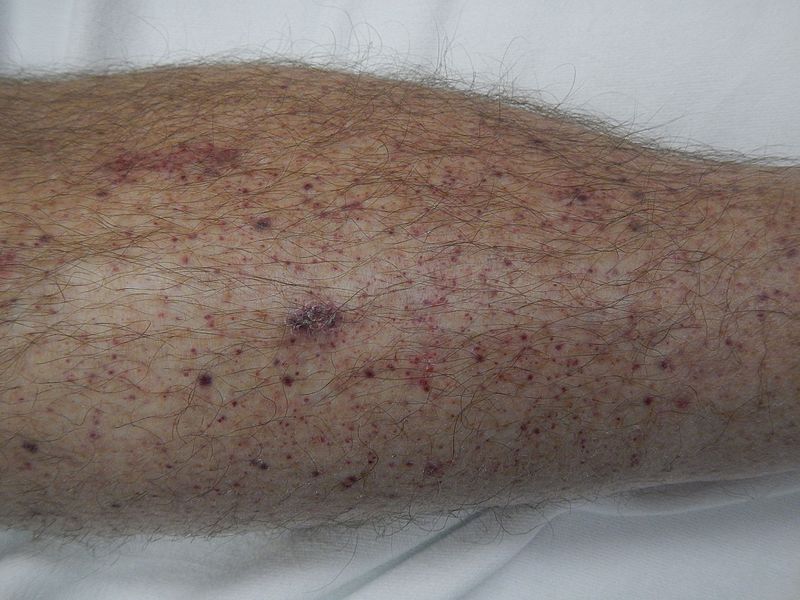
Petéquias na perna de um paciente com púrpura trombocitopénica imune
Imagem: “Petechiae on lower leg” por James Heilman, MD. Licença: CC BY-SA 4.0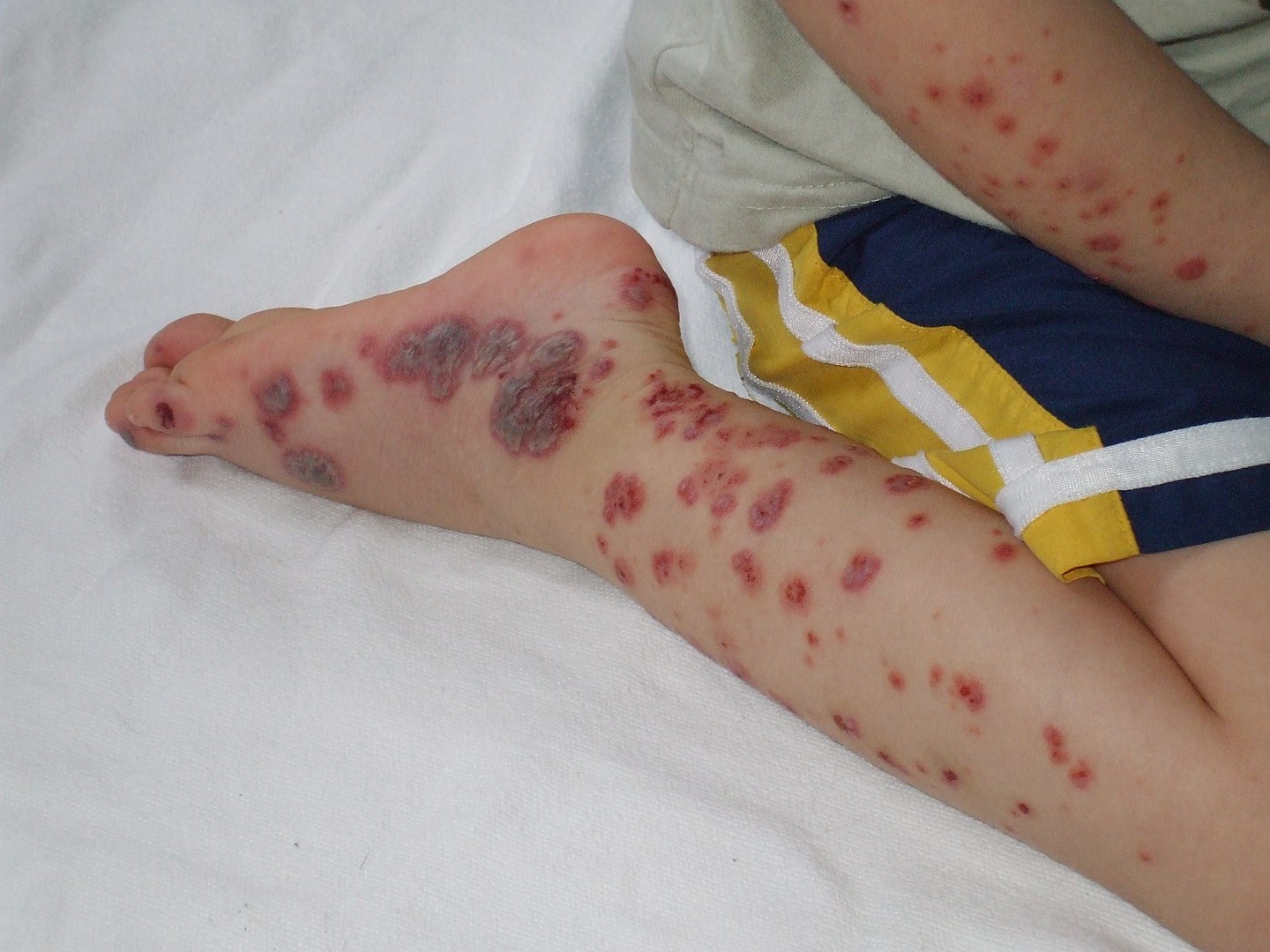
Púrpura evidente na extremidade distal do membro inferior de um paciente pediátrico
Imagem: “Purpura2” por Okwikikim. Licença: Domínio PúblicoO diagnóstico da trombocitopenia em si é feito com exames laboratoriais. Podem obter-se pistas acerca da etiologia subjacente a partir da história clínica e do exame objetivo, e exames laboratoriais adicionais podem auxiliar ainda mais MAIS Androgen Insensitivity Syndrome no diagnóstico.
Os seguintes exames devem ser solicitados em todos os pacientes com trombocitopenia:
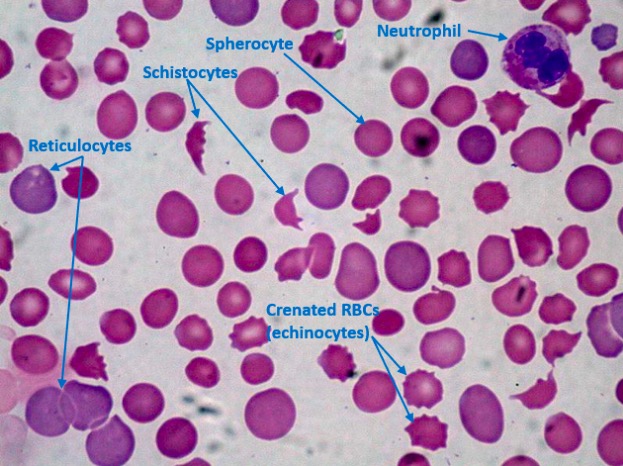
Esfregaço de sangue periférico em paciente com CID mostrando trombocitopenia, esquizócitos e esferócitos:
Estão presentes muitos reticulócitos (eritrócitos imaturos), indicando uma resposta hematopoiética rápida da medula óssea. As contagens de plaquetas são reduzidas (nenhuma é vista aqui) por causa do consumo devido à CID. Normalmente, deve haver pelo menos 8 plaquetas num único campo de imersão em óleo de 1000×; 1 plaqueta indica aproximadamente 20.000 plaquetas/µL no sangue periférico.
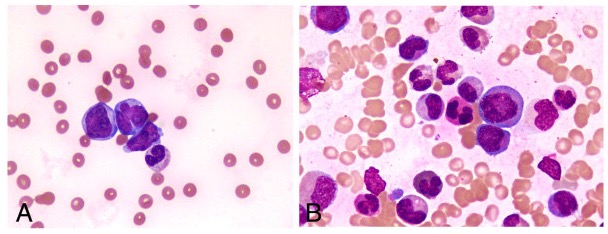
Esfregaços periféricos mostrando uma escassez de plaquetas
Imagem: “Peripheral blood smear and bone marrow aspirate” por Shabneez Hussain. Licença: CC BY 2.0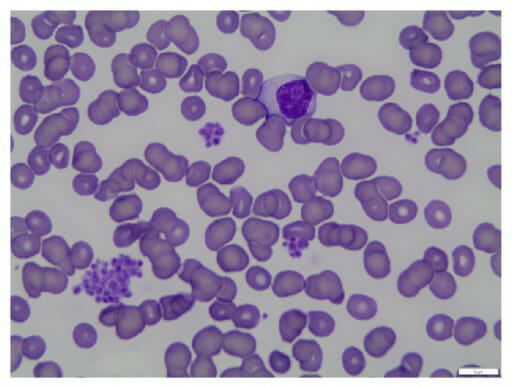
Esfregaço sanguíneo com pseudotrombocitopenia: A pseudotrombocitopenia é causada pela agregação de plaquetas in vitro devido à aglutinação.
Imagem: “fig1: Peripheral blood smear (100x oil)” por Geok Chin Tan. Licença: CC BY 4.0Os exames a seguir devem ser solicitados somente conforme indicação clínica para ajudar a identificar a etiologia subjacente.
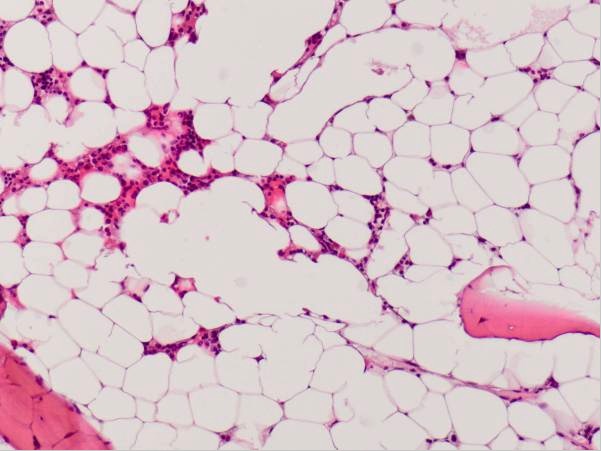
Medula óssea hipocelular em paciente com anemia aplásica
Imagem: “Hypocellular bone marrow showing only little hematopoiesis” por Department of Obstetrics of Gynaecology, Erasmus University Medical Center, PO Box 2040, 3000 CA Rotterdam, The Netherlands. Licença: CC BY 2.0O tratamento da trombocitopenia depende principalmente da abordagem da causa subjacente, da monitorização da contagem de plaquetas e da prevenção/controlo de todas as complicações hemorrágicas.
