Primary myelofibrosis (PMF) is a myeloproliferative neoplasm characterized by chronic myeloproliferation with nonclonal fibroblastic deposition, resulting in bone marrow Bone marrow The soft tissue filling the cavities of bones. Bone marrow exists in two types, yellow and red. Yellow marrow is found in the large cavities of large bones and consists mostly of fat cells and a few primitive blood cells. Red marrow is a hematopoietic tissue and is the site of production of erythrocytes and granular leukocytes. Bone marrow is made up of a framework of connective tissue containing branching fibers with the frame being filled with marrow cells. Bone Marrow: Composition and Hematopoiesis fibrosis Fibrosis Any pathological condition where fibrous connective tissue invades any organ, usually as a consequence of inflammation or other injury. Bronchiolitis Obliterans. The abnormality stems from genetic mutations Genetic Mutations Carcinogenesis of the hematopoietic stem cells Hematopoietic stem cells Progenitor cells from which all blood cells derived. They are found primarily in the bone marrow and also in small numbers in the peripheral blood. Bone Marrow: Composition and Hematopoiesis (most commonly involving the driver mutations JAK2, CALR, or MPL). Primary symptoms are anemia Anemia Anemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types and extramedullary hematopoiesis Hematopoiesis The development and formation of various types of blood cells. Hematopoiesis can take place in the bone marrow (medullary) or outside the bone marrow (extramedullary hematopoiesis). Bone Marrow: Composition and Hematopoiesis, which include severe fatigue Fatigue The state of weariness following a period of exertion, mental or physical, characterized by a decreased capacity for work and reduced efficiency to respond to stimuli. Fibromyalgia, weight loss Weight loss Decrease in existing body weight. Bariatric Surgery, and hepatosplenomegaly Hepatosplenomegaly Cytomegalovirus. Laboratory findings include anemia Anemia Anemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types (and other cytopenias Cytopenias IPEX Syndrome), and bone marrow Bone marrow The soft tissue filling the cavities of bones. Bone marrow exists in two types, yellow and red. Yellow marrow is found in the large cavities of large bones and consists mostly of fat cells and a few primitive blood cells. Red marrow is a hematopoietic tissue and is the site of production of erythrocytes and granular leukocytes. Bone marrow is made up of a framework of connective tissue containing branching fibers with the frame being filled with marrow cells. Bone Marrow: Composition and Hematopoiesis biopsy Biopsy Removal and pathologic examination of specimens from the living body. Ewing Sarcoma reveals extensive fibrosis Fibrosis Any pathological condition where fibrous connective tissue invades any organ, usually as a consequence of inflammation or other injury. Bronchiolitis Obliterans. Management is with allogeneic hematopoietic stem cell transplantation Hematopoietic stem cell transplantation Transfer of hematopoietic stem cells from bone marrow or blood between individuals within the same species (homologous transplantation) or transfer within the same individual (autologous transplantation). Hematopoietic stem cell transplantation has been used as an alternative to bone marrow transplantation in the treatment of a variety of neoplasms. Organ Transplantation and symptom-directed interventions.
Last updated: Dec 15, 2025
Primary myelofibrosis (PMF) is a chronic myeloproliferative neoplasm characterized by proliferation of myeloid cells Myeloid Cells The classes of bone marrow-derived blood cells in the monocytic series (monocytes and their precursors) and granulocytic series (granulocytes and their precursors). White Myeloid Cells: Histology, with nonclonal fibroblast proliferation and hyperactivity Hyperactivity Attention Deficit Hyperactivity Disorder, resulting in an obliterative marrow fibrosis Fibrosis Any pathological condition where fibrous connective tissue invades any organ, usually as a consequence of inflammation or other injury. Bronchiolitis Obliterans. Other names include chronic idiopathic Idiopathic Dermatomyositis myelofibrosis and agnogenic myeloid metaplasia Metaplasia A condition in which there is a change of one adult cell type to another similar adult cell type. Cellular Adaptation.
Hematopoiesis Hematopoiesis The development and formation of various types of blood cells. Hematopoiesis can take place in the bone marrow (medullary) or outside the bone marrow (extramedullary hematopoiesis). Bone Marrow: Composition and Hematopoiesis starts with the hematopoietic stem cell, which is prompted to divide and differentiate with appropriate chemical stimuli (hemopoietic growth factors)
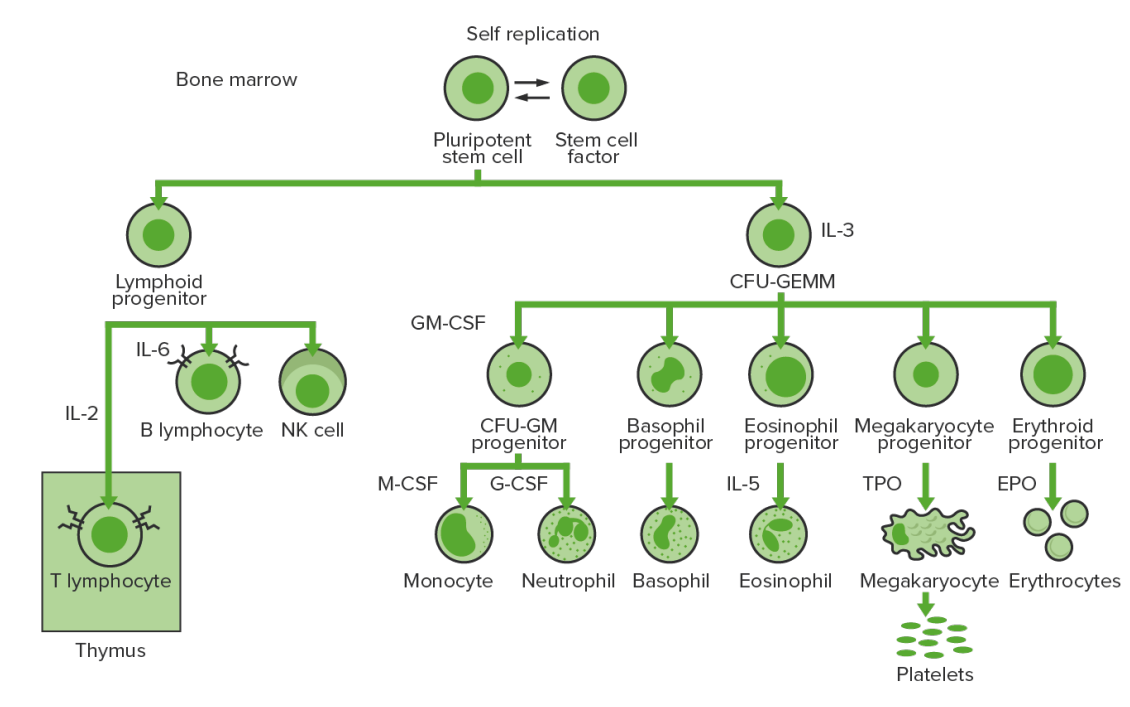
Bone-marrow hematopoiesis: proliferation and differentiation of the formed elements of blood.
CFU-GEMM: colony-forming unit–granulocyte, erythrocyte, monocyte, megakaryocyte
CFU-GM: colony-forming unit–granulocyte-macrophage
GM-CSF: granulocyte-macrophage colony-stimulating factor
M-CSF: macrophage colony-stimulating factor
G-CSF: granulocyte colony-stimulating factor
NK: natural killer
TPO: thrombopoietin
Mutations in genes Genes A category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms. DNA Types and Structure associated with hematopoiesis Hematopoiesis The development and formation of various types of blood cells. Hematopoiesis can take place in the bone marrow (medullary) or outside the bone marrow (extramedullary hematopoiesis). Bone Marrow: Composition and Hematopoiesis are noted in a significant proportion of cases of PMF.
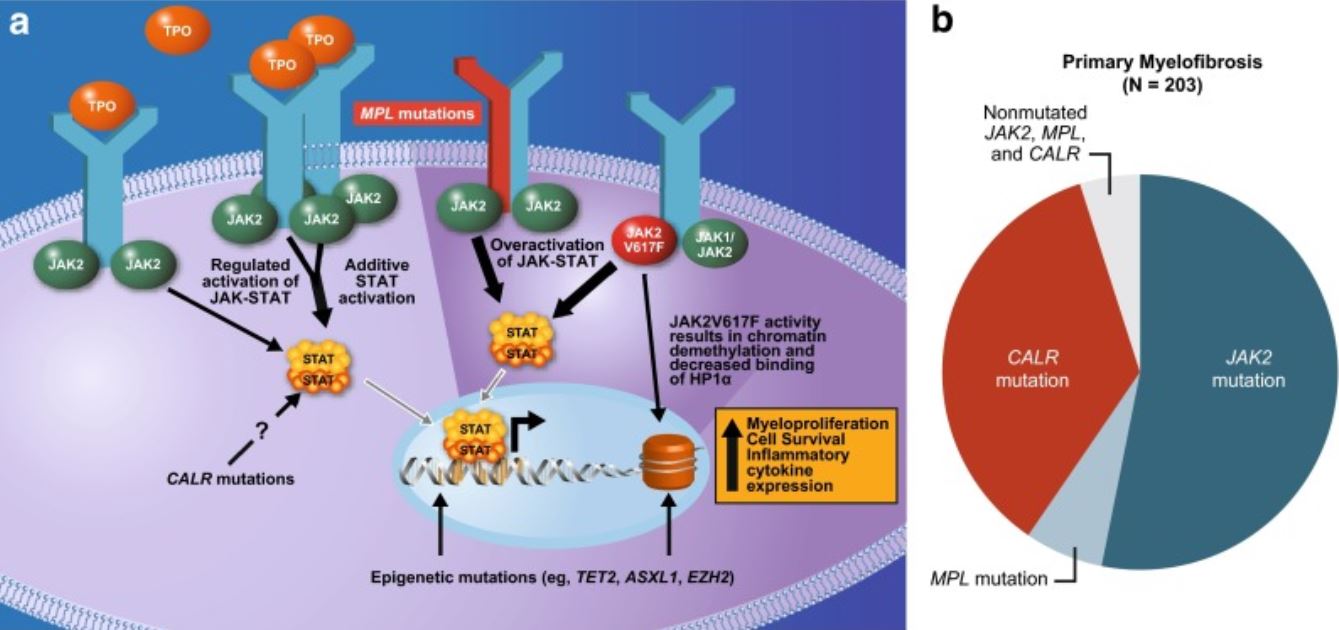
Mutations underlying the pathophysiology of myelofibrosis:
Commonly affects the JAK–STAT pathway
JAK2 (JAK2 gene involved): non-receptor tyrosine kinase facilitating extracellular signals to reach the nucleus and activate genes.
MPL or thrombopoietin receptor (MPL gene involved): activation of the thrombopoietin receptor
Calreticulin (CALR gene involved) is a regulator of STAT, and mutation causes activation of thrombopoietin receptor.
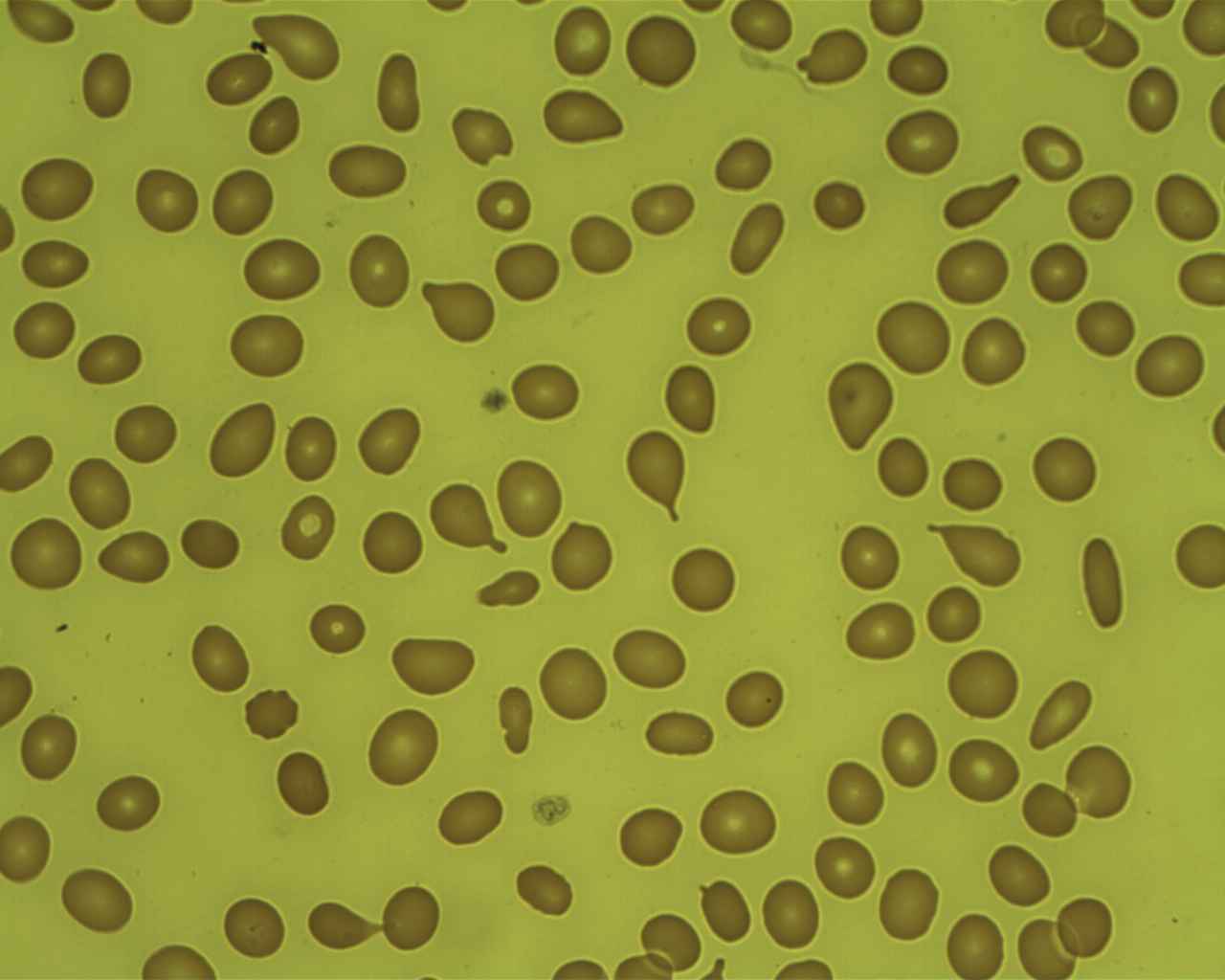
Myelofibrosis: teardrop cells in peripheral blood smear
Image: “Tear Drop Poikilocytes” by Prof. Osaro Erhabor. License: CC0 1.0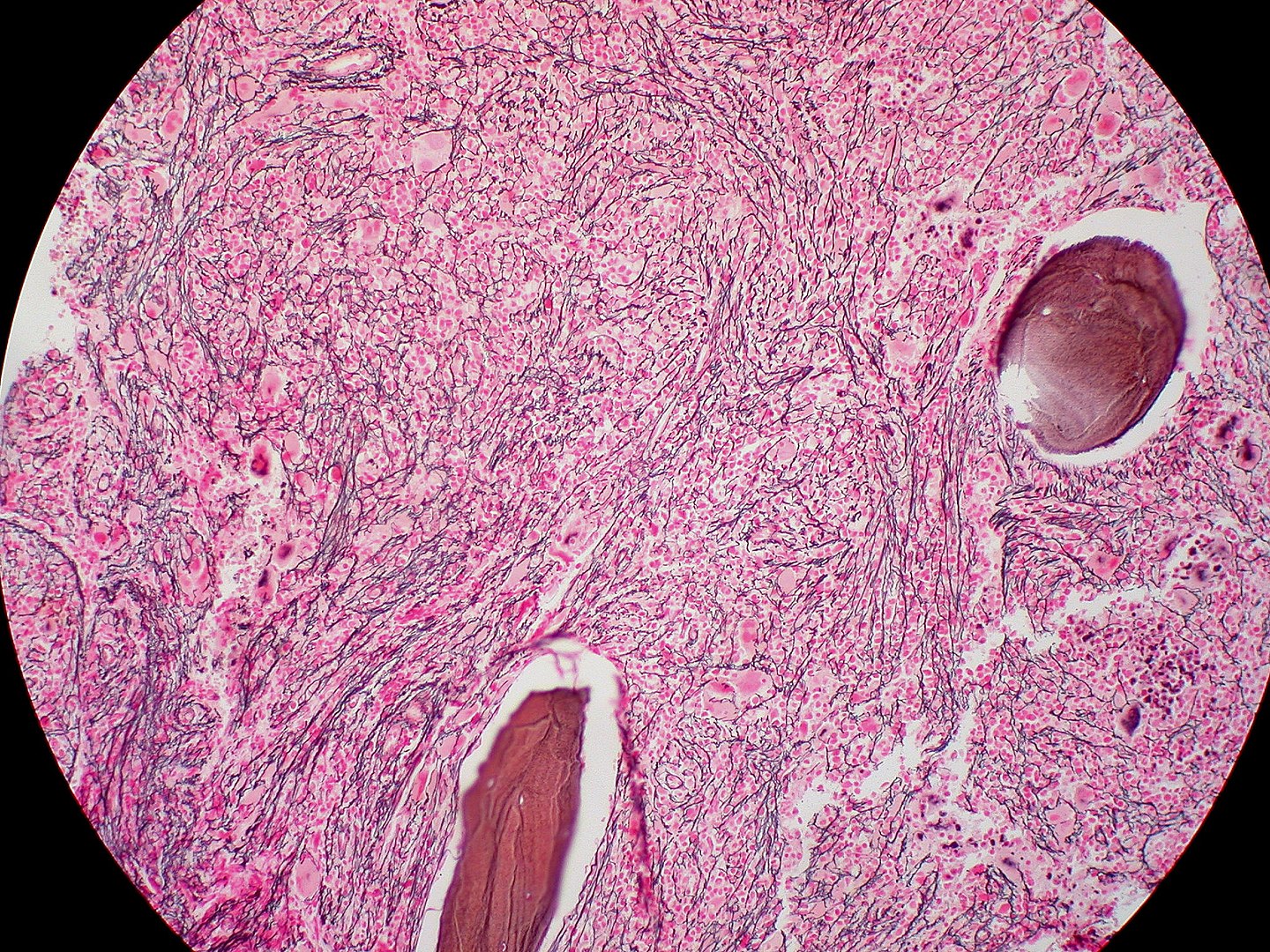
Bone marrow biopsy of myelofibrosis: extensive fibrosis (reticulin stain)
Image: “Myelofibrosis, Reticulin Stain (6032644716)” by Ed Uthman. License: CC BY 2.0To remember PMF in summary: Bone marrow Bone marrow The soft tissue filling the cavities of bones. Bone marrow exists in two types, yellow and red. Yellow marrow is found in the large cavities of large bones and consists mostly of fat cells and a few primitive blood cells. Red marrow is a hematopoietic tissue and is the site of production of erythrocytes and granular leukocytes. Bone marrow is made up of a framework of connective tissue containing branching fibers with the frame being filled with marrow cells. Bone Marrow: Composition and Hematopoiesis is crying (teardrop cells) because it is fibrosed and is a dry tap Dry Tap Hairy Cell Leukemia.
Pre-fibrotic/early primary myelofibrosis (pre-PMF)
Overt (fibrotic) primary myelofibrosis
Myeloproliferative neoplasms Neoplasms New abnormal growth of tissue. Malignant neoplasms show a greater degree of anaplasia and have the properties of invasion and metastasis, compared to benign neoplasms. Benign Bone Tumors can be compared with the following WHO classification:
| Disease | Mutations | Key points |
|---|---|---|
| CML CML Chronic myeloid leukemia is a malignant proliferation of the granulocytic cell line characterized by a fairly normal differentiation. The underlying genetic abnormality is the Philadelphia chromosome, an abbreviated chromosome 22, resulting from reciprocal (9;22)(q34;q11) translocation. Chronic Myeloid Leukemia | BCR-ABL1 ( Philadelphia chromosome Philadelphia chromosome An aberrant form of human chromosome 22 characterized by translocation of the distal end of chromosome 9 from 9q34, to the long arm of chromosome 22 at 22q11. It is present in the bone marrow cells of 80 to 90 percent of patients with chronic myelocytic leukemia. Acute Lymphoblastic Leukemia) | Proliferation of mature and maturing granulocytes Granulocytes Leukocytes with abundant granules in the cytoplasm. They are divided into three groups according to the staining properties of the granules: neutrophilic, eosinophilic, and basophilic. Mature granulocytes are the neutrophils; eosinophils; and basophils. White Myeloid Cells: Histology |
| ET | JAK2, CALR, or MPL | Excessive clonal platelet production |
| Polycythemia Polycythemia An increase in the total red cell mass of the blood. Renal Cell Carcinoma vera ( PV PV Polycythemia vera (PV) is a chronic myeloproliferative neoplasm characterized by the overproduction of rbcs. In addition, the wbc and platelet counts are also increased, which differentiate pv from erythrocytosis seen with chronic hypoxia and other chronic conditions. Polycythemia Vera) | JAK2 | Elevated RBC mass Mass Three-dimensional lesion that occupies a space within the breast Imaging of the Breast |
| PMF | JAK2, CALR, or MPL | Obliterative bone marrow Bone marrow The soft tissue filling the cavities of bones. Bone marrow exists in two types, yellow and red. Yellow marrow is found in the large cavities of large bones and consists mostly of fat cells and a few primitive blood cells. Red marrow is a hematopoietic tissue and is the site of production of erythrocytes and granular leukocytes. Bone marrow is made up of a framework of connective tissue containing branching fibers with the frame being filled with marrow cells. Bone Marrow: Composition and Hematopoiesis fibrosis Fibrosis Any pathological condition where fibrous connective tissue invades any organ, usually as a consequence of inflammation or other injury. Bronchiolitis Obliterans |
Other types:
Assessment of risk is important in making treatment decisions.