Essential thrombocythemia (ET) is a type of myeloproliferative neoplasm characterized by the clonal thrombocytosis linked to somatic mutations involving Janus kinase 2 (JAK2), calreticulin (CALR), and myeloproliferative leukemia virus Virus Viruses are infectious, obligate intracellular parasites composed of a nucleic acid core surrounded by a protein capsid. Viruses can be either naked (non-enveloped) or enveloped. The classification of viruses is complex and based on many factors, including type and structure of the nucleoid and capsid, the presence of an envelope, the replication cycle, and the host range. Virology oncogene (MPL). Patients Patients Individuals participating in the health care system for the purpose of receiving therapeutic, diagnostic, or preventive procedures. Clinician–Patient Relationship can be asymptomatic or present with vasomotor symptoms such as headaches, erythromelalgia Erythromelalgia A peripheral arterial disease that is characterized by the triad of erythema, burning pain, and increased skin temperature of the extremities (or red, painful extremities). Erythromelalgia may be classified as primary or idiopathic, familial or non-familial. Secondary erythromelalgia is associated with other diseases, the most common being myeloproliferative disorders. Polycythemia Vera, and transient visual disturbances. The clinical course can be complicated by thrombohemorrhagic events as well as progression to myelofibrosis and AML AML Acute myeloid leukemia (AML) is a hematologic malignancy characterized by the uncontrolled proliferation of myeloid precursor cells. Seen predominantly in older adults, AML includes an accumulation of myeloblasts and a replacement of normal marrow by malignant cells, which leads to impaired hematopoiesis. Acute Myeloid Leukemia. The diagnosis is based on a laboratory finding of thrombocytosis, bone marrow Bone marrow The soft tissue filling the cavities of bones. Bone marrow exists in two types, yellow and red. Yellow marrow is found in the large cavities of large bones and consists mostly of fat cells and a few primitive blood cells. Red marrow is a hematopoietic tissue and is the site of production of erythrocytes and granular leukocytes. Bone marrow is made up of a framework of connective tissue containing branching fibers with the frame being filled with marrow cells. Bone Marrow: Composition and Hematopoiesis biopsy Biopsy Removal and pathologic examination of specimens from the living body. Ewing Sarcoma, and genetic studies. Treatment aims to reduce platelet count by cytoreductive agents ( hydroxyurea Hydroxyurea An antineoplastic agent that inhibits DNA synthesis through the inhibition of ribonucleoside diphosphate reductase. Antimetabolite Chemotherapy) and to decrease thrombosis Thrombosis Formation and development of a thrombus or blood clot in the blood vessel. Epidemic Typhus with aspirin Aspirin The prototypical analgesic used in the treatment of mild to moderate pain. It has anti-inflammatory and antipyretic properties and acts as an inhibitor of cyclooxygenase which results in the inhibition of the biosynthesis of prostaglandins. Aspirin also inhibits platelet aggregation and is used in the prevention of arterial and venous thrombosis. Nonsteroidal Antiinflammatory Drugs (NSAIDs) and systemic anticoagulation Anticoagulation Pulmonary Hypertension Drugs based on thrombosis Thrombosis Formation and development of a thrombus or blood clot in the blood vessel. Epidemic Typhus risk stratification.
Last updated: Jun 30, 2026
Essential thrombocythemia (ET) is a chronic myeloproliferative neoplasm characterized by excessive platelet production and increased thrombotic and hemorrhagic tendency. Other names are essential thrombocytosis and primary thrombocytosis.
Hematopoiesis Hematopoiesis The development and formation of various types of blood cells. Hematopoiesis can take place in the bone marrow (medullary) or outside the bone marrow (extramedullary hematopoiesis). Bone Marrow: Composition and Hematopoiesis starts with the hematopoietic stem cell, which is prompted to divide and differentiate with appropriate chemical stimuli (hemopoietic growth factors).
Production of platelets Platelets Platelets are small cell fragments involved in hemostasis. Thrombopoiesis takes place primarily in the bone marrow through a series of cell differentiation and is influenced by several cytokines. Platelets are formed after fragmentation of the megakaryocyte cytoplasm. Platelets: Histology:
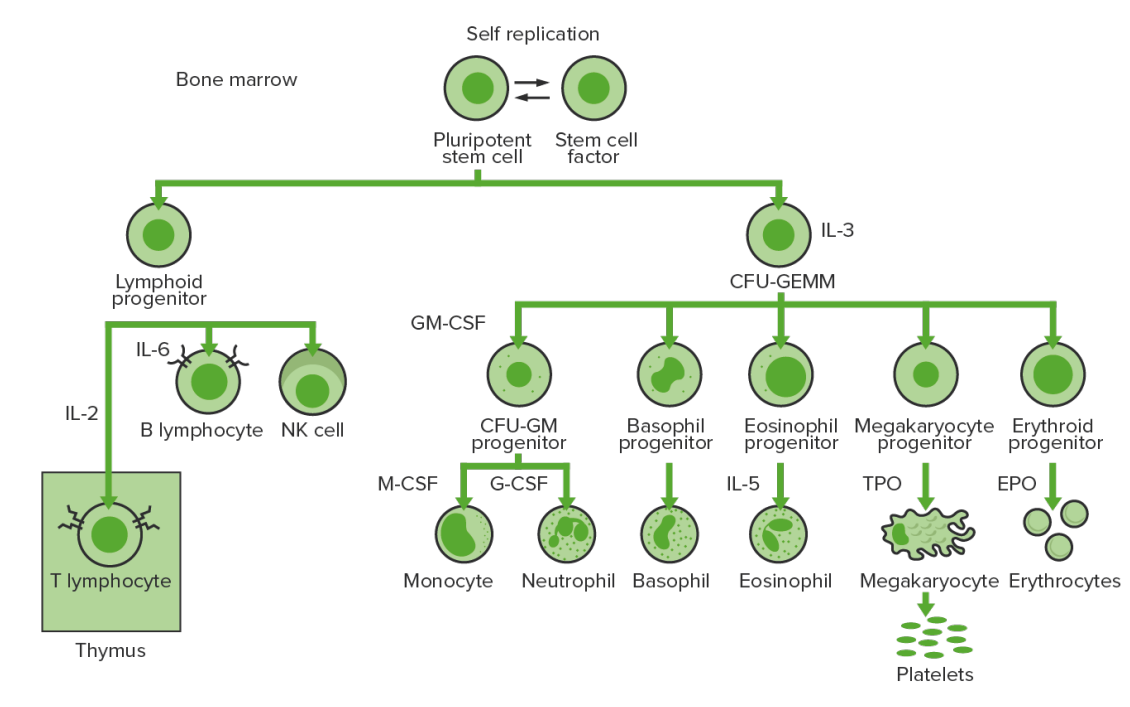
Bone-marrow hematopoiesis: proliferation and differentiation of the formed elements of blood.
CFU-GEMM: colony-forming unit–granulocyte, erythrocyte, monocyte, megakaryocyte
CFU-GM: colony-forming unit–granulocyte-macrophage
GM-CSF: granulocyte-macrophage colony-stimulating factor
M-CSF: macrophage colony-stimulating factor
G-CSF: granulocyte colony-stimulating factor
NK: natural killer
TPO: thrombopoietin
Increased incidence Incidence The number of new cases of a given disease during a given period in a specified population. It also is used for the rate at which new events occur in a defined population. It is differentiated from prevalence, which refers to all cases in the population at a given time. Measures of Disease Frequency due to:
Increased hemorrhagic diathesis in ET is due to:
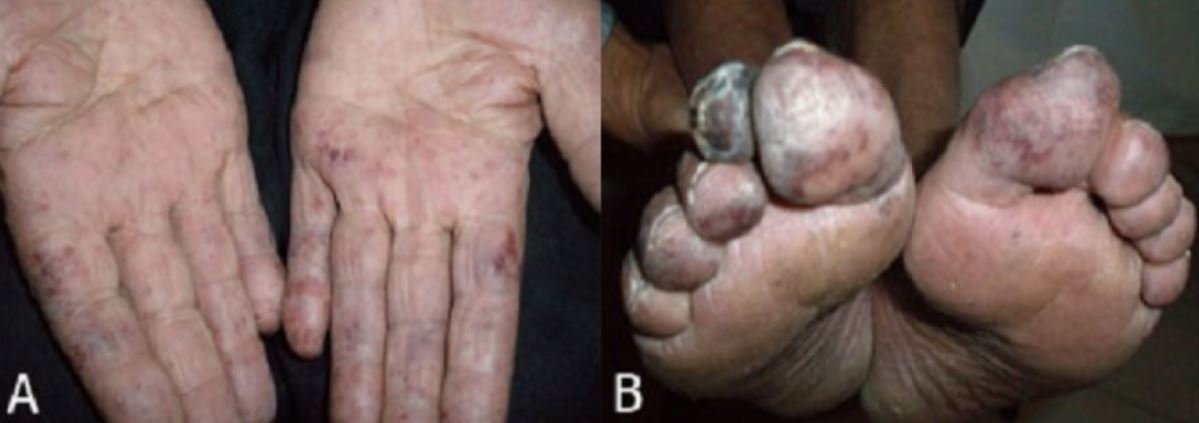
Physical findings of essential thrombocythemia:
Livedo reticularis and areas of mottled bluish ischemia of fingers (A) and toe gangrene (B)
Diagnosis requires all 4 major criteria or first 3 major criteria and 1 minor criterion.
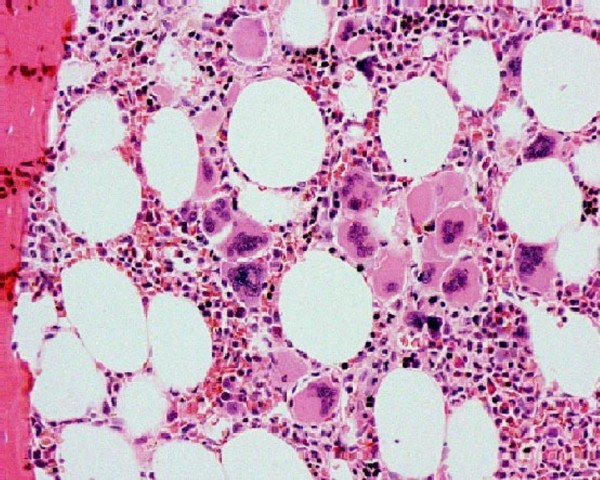
Bone marrow biopsy in essential thrombocythemia:
Increased megakaryocytes can be seen.
Myeloproliferative neoplasms Neoplasms New abnormal growth of tissue. Malignant neoplasms show a greater degree of anaplasia and have the properties of invasion and metastasis, compared to benign neoplasms. Benign Bone Tumors can be compared with the following WHO classification:
| Disease | Mutations | Key points |
|---|---|---|
| CML CML Chronic myeloid leukemia is a malignant proliferation of the granulocytic cell line characterized by a fairly normal differentiation. The underlying genetic abnormality is the Philadelphia chromosome, an abbreviated chromosome 22, resulting from reciprocal (9;22)(q34;q11) translocation. Chronic Myeloid Leukemia | BCR-ABL1 ( Philadelphia chromosome Philadelphia chromosome An aberrant form of human chromosome 22 characterized by translocation of the distal end of chromosome 9 from 9q34, to the long arm of chromosome 22 at 22q11. It is present in the bone marrow cells of 80 to 90 percent of patients with chronic myelocytic leukemia. Acute Lymphoblastic Leukemia) | Proliferation of mature and maturing granulocytes Granulocytes Leukocytes with abundant granules in the cytoplasm. They are divided into three groups according to the staining properties of the granules: neutrophilic, eosinophilic, and basophilic. Mature granulocytes are the neutrophils; eosinophils; and basophils. White Myeloid Cells: Histology |
| ET | JAK2, CALR, or MPL | Excessive clonal platelet production |
| Polycythemia Polycythemia An increase in the total red cell mass of the blood. Renal Cell Carcinoma vera ( PV PV Polycythemia vera (PV) is a chronic myeloproliferative neoplasm characterized by the overproduction of rbcs. In addition, the wbc and platelet counts are also increased, which differentiate pv from erythrocytosis seen with chronic hypoxia and other chronic conditions. Polycythemia Vera) | JAK2 | Elevated RBC mass Mass Three-dimensional lesion that occupies a space within the breast Imaging of the Breast |
| Primary myelofibrosis Primary myelofibrosis Primary myelofibrosis (PMF) is a myeloproliferative neoplasm characterized by chronic myeloproliferation with nonclonal fibroblastic deposition, resulting in bone marrow fibrosis. The abnormality stems from genetic mutations of the hematopoietic stem cells (typically, JAK2 mutation). Primary symptoms are anemia and extramedullary hematopoiesis,. Primary Myelofibrosis (PMF) | JAK2, CALR, or MPL | Obliterative bone marrow fibrosis Bone Marrow Fibrosis Primary Myelofibrosis |
Other types:
| Risk for thrombosis Thrombosis Formation and development of a thrombus or blood clot in the blood vessel. Epidemic Typhus | Features | Treatment |
|---|---|---|
| Very low risk |
|
|
| Low risk |
|
|
| Intermediate risk |
|
|
| High risk |
|
|