La trombocitemia esencial es un tipo de neoplasia mieloproliferativa caracterizada por la trombocitosis clonal vinculada a mutaciones somáticas que involucran a Janus quinasa 2, calreticulina y el oncogén del virus Virus Viruses are infectious, obligate intracellular parasites composed of a nucleic acid core surrounded by a protein capsid. Viruses can be either naked (non-enveloped) or enveloped. The classification of viruses is complex and based on many factors, including type and structure of the nucleoid and capsid, the presence of an envelope, the replication cycle, and the host range. Virology de la leucemia mieloproliferativa. Los LOS Neisseria pacientes pueden estar asintomáticos o presentar síntomas vasomotores como cefaleas, eritromelalgia y alteraciones visuales transitorias. La evolución puede complicarse con eventos trombohemorrágicos, así como con la progresión a mielofibrosis y LMA. El diagnóstico se basa en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum un hallazgo de laboratorio de trombocitosis, biopsia de médula ósea y estudios genéticos. El tratamiento tiene como objetivo reducir el recuento de plaquetas con agentes citorreductores (hidroxiurea) y disminuir la trombosis con aspirina y anticoagulación sistémica según la estratificación del riesgo de trombosis.
Last updated: Jun 30, 2026
La trombocitemia esencial es una neoplasia mieloproliferativa crónica caracterizada por una producción excesiva de plaquetas y un aumento de la tendencia trombótica y hemorrágica. Otros nombres son trombocitosis esencial y trombocitosis primaria.
La hematopoyesis comienza con la célula madre hematopoyética, que se incita a dividirse y diferenciarse con estímulos químicos apropiados (factores de crecimiento hematopoyético).
Producción de plaquetas:
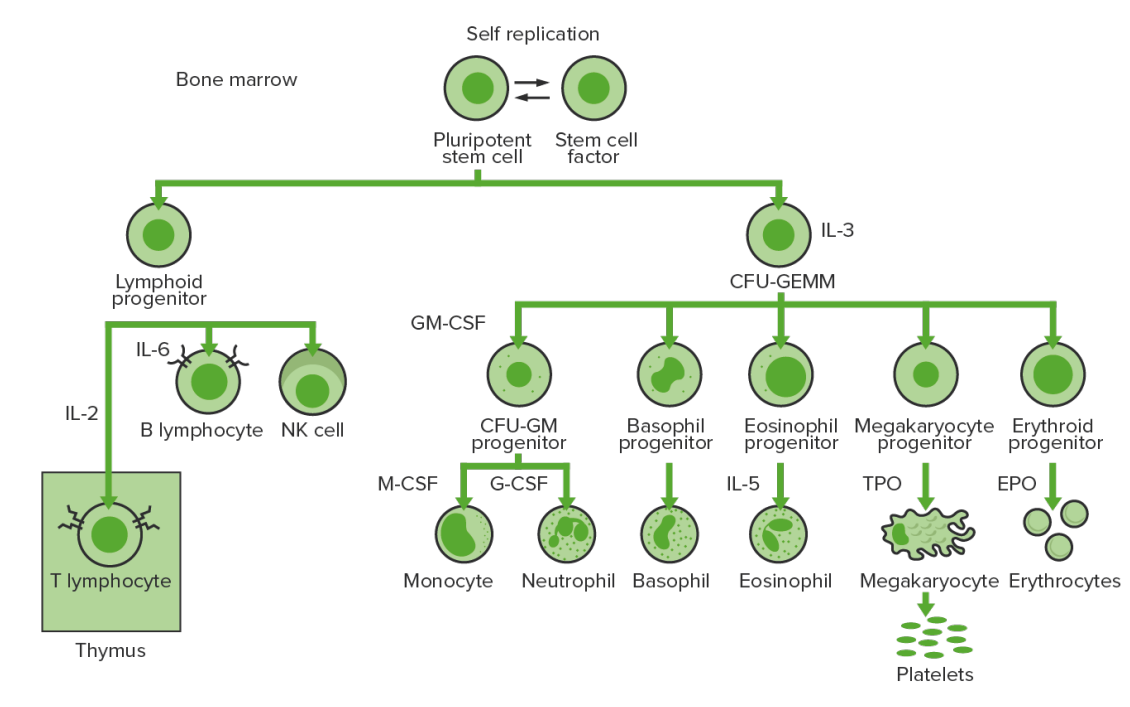
Hematopoyesis de la médula ósea: proliferación y diferenciación de los elementos sólidos de la sangre.
CFU-GEMM: unidad formadora de colonias: granulocitos, eritrocitos, monocitos, megacariocitos
CFU-GM: unidad formadora de colonias-granulocitos-macrófagos
GM-CSF: factor estimulante de colonias de granulocitos y macrófagos
M-CSF: factor estimulante de colonias de macrófagos
G-CSF: factor estimulante de colonias de granulocitos
NK: asesina natural
TPO: trombopoyetina
Mayor incidencia debido a:
El aumento de la diátesis hemorrágica se debe a:
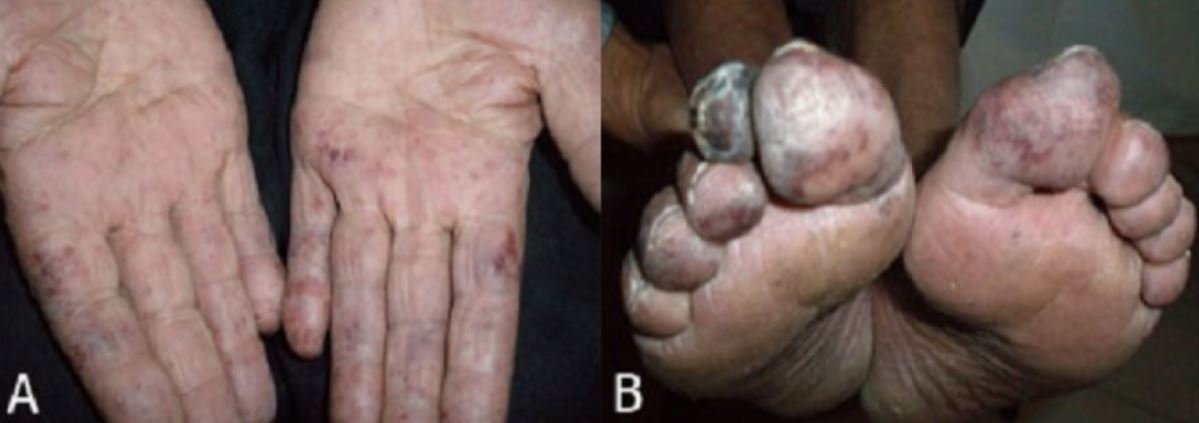
Hallazgos físicos de trombocitemia esencial:
Livedo reticularis y áreas de isquemia azulada moteada de los dedos (A) y gangrena del dedo del pie (B)
El diagnóstico requiere los LOS Neisseria 4 criterios mayores o los LOS Neisseria primeros 3 criterios mayores y 1 criterio menor.
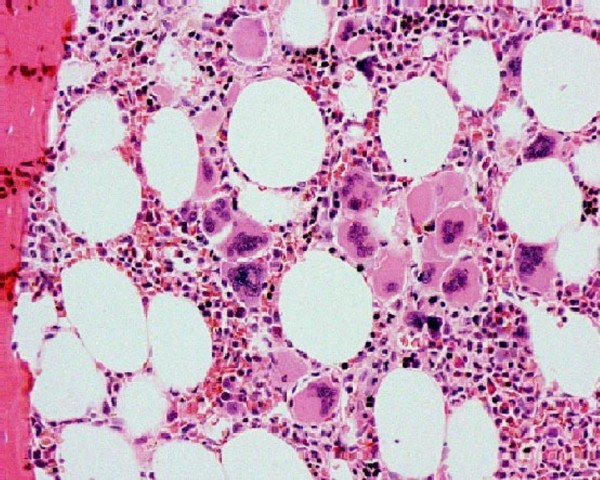
Biopsia de médula ósea en trombocitemia esencial:
Se puede observar un aumento de megacariocitos.
Las neoplasias mieloproliferativas se pueden comparar con la siguiente clasificación de la WHO:
| Enfermedad | Mutaciones | Puntos clave |
|---|---|---|
| LMC | BCR-ABL1 (cromosoma Filadelfia) | Proliferación de granulocitos maduros y en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum proceso de maduración. |
| Trombocitemia esencial | JAK2, CALR o leucemia mieloproliferativa | Producción clonal excesiva de plaquetas |
| Policitemia vera | JAK2 | Conteo elevado de eritrocitos |
| Mielofibrosis primaria | JAK2, CALR o leucemia mieloproliferativa | Fibrosis Fibrosis Any pathological condition where fibrous connective tissue invades any organ, usually as a consequence of inflammation or other injury. Bronchiolitis Obliterans obliterante de la médula ósea |
Otros tipos:
| Riesgo de trombosis | Características | Tratamiento |
|---|---|---|
| Muy bajo riesgo |
|
|
| Riesgo bajo |
|
|
| Riesgo intermedio |
|
|
| Alto riesgo |
|
|