Chronic myeloid leukemia is a malignant proliferation of the granulocytic cell line characterized by a fairly normal differentiation. The underlying genetic abnormality is the Philadelphia chromosome Philadelphia chromosome An aberrant form of human chromosome 22 characterized by translocation of the distal end of chromosome 9 from 9q34, to the long arm of chromosome 22 at 22q11. It is present in the bone marrow cells of 80 to 90 percent of patients with chronic myelocytic leukemia. Acute Lymphoblastic Leukemia, an abbreviated chromosome Chromosome In a prokaryotic cell or in the nucleus of a eukaryotic cell, a structure consisting of or containing DNA which carries the genetic information essential to the cell. Basic Terms of Genetics 22, resulting from reciprocal (9;22)(q34;q11) translocation. The chromosome Chromosome In a prokaryotic cell or in the nucleus of a eukaryotic cell, a structure consisting of or containing DNA which carries the genetic information essential to the cell. Basic Terms of Genetics contains the BCR-ABL1 fusion gene Gene A category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms. Basic Terms of Genetics (from ABL1 on chromosome 9 Chromosome 9 Friedreich Ataxia and BCR BCR Lymphocytes: Histology on chromosome Chromosome In a prokaryotic cell or in the nucleus of a eukaryotic cell, a structure consisting of or containing DNA which carries the genetic information essential to the cell. Basic Terms of Genetics 22), which induces constitutive tyrosine Tyrosine A non-essential amino acid. In animals it is synthesized from phenylalanine. It is also the precursor of epinephrine; thyroid hormones; and melanin. Synthesis of Nonessential Amino Acids kinase activation and consequently, uncontrolled granulocyte production. Patients Patients Individuals participating in the health care system for the purpose of receiving therapeutic, diagnostic, or preventive procedures. Clinician–Patient Relationship with CML can be asymptomatic or have constitutional symptoms Constitutional Symptoms Antineutrophil Cytoplasmic Antibody (ANCA)-Associated Vasculitis, sternal pain Pain An unpleasant sensation induced by noxious stimuli which are detected by nerve endings of nociceptive neurons. Pain: Types and Pathways, and splenomegaly Splenomegaly Splenomegaly is pathologic enlargement of the spleen that is attributable to numerous causes, including infections, hemoglobinopathies, infiltrative processes, and outflow obstruction of the portal vein. Splenomegaly. Laboratory studies show an elevated WBC, and a peripheral blood smear Peripheral Blood Smear Anemia: Overview and Types shows increased numbers of immature cells. Demonstration of Philadelphia translocation using cytogenetic techniques is considered the gold standard diagnostic test. Without treatment, CML generally has a triphasic course progressing from a chronic phase Chronic phase HIV Infection and AIDS to an accelerated phase and leading to a terminal blast crisis. Management includes therapy with tyrosine Tyrosine A non-essential amino acid. In animals it is synthesized from phenylalanine. It is also the precursor of epinephrine; thyroid hormones; and melanin. Synthesis of Nonessential Amino Acids kinase inhibitors and palliative agents, as well as allogeneic hematopoietic cell transplantation.
Last updated: May 17, 2024
Chronic myeloid leukemia is a chronic myeloproliferative neoplasm characterized by uncontrolled and dysregulated proliferation of the granulocytic lineage (mature and maturing cells), with a maintained capacity for differentiation. Chronic myeloid leukemia is also known as chronic myelocytic leukemia or chronic myelogenous leukemia.
Hematopoiesis Hematopoiesis The development and formation of various types of blood cells. Hematopoiesis can take place in the bone marrow (medullary) or outside the bone marrow (extramedullary hematopoiesis). Bone Marrow: Composition and Hematopoiesis starts with a hematopoietic stem cell, which is prompted to divide and differentiate with appropriate chemical stimuli (hemopoietic growth factors).
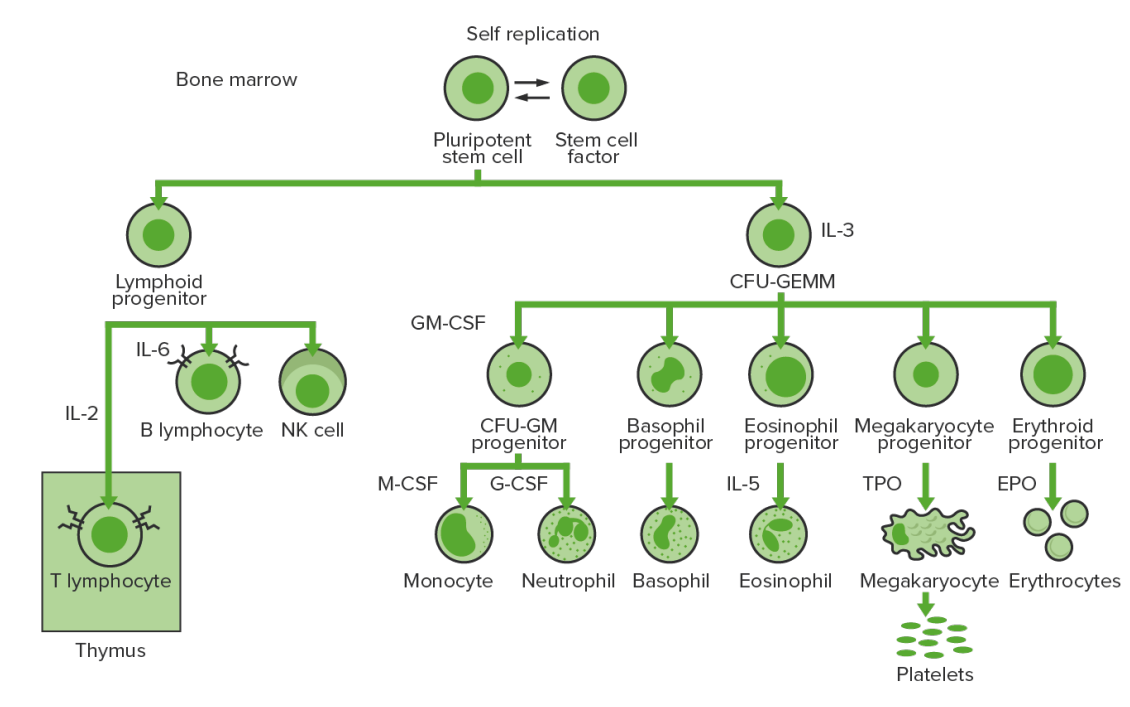
Bone marrow hematopoiesis:
Proliferation and differentiation of the formed elements of blood:
In CML, there is sustained proliferation of cells in the granulocytic line (myeloblasts → neutrophils, basophils, eosinophils). Both mature and maturing cells are seen; thus, there are cells that are only partially effective.
CFU-GEMM: colony-forming unit–granulocyte, erythrocyte, monocyte, megakaryocyte
GM-CSF: granulocyte macrophage colony-stimulating factor
M-CSF: macrophage colony-stimulating factor
G-CSF: granulocyte colony-stimulating factor
NK: natural killer
Chronic myeloid leukemia is an acquired abnormality of the hematopoietic stem cells Hematopoietic stem cells Progenitor cells from which all blood cells derived. They are found primarily in the bone marrow and also in small numbers in the peripheral blood. Bone Marrow: Composition and Hematopoiesis in the bone marrow Bone marrow The soft tissue filling the cavities of bones. Bone marrow exists in two types, yellow and red. Yellow marrow is found in the large cavities of large bones and consists mostly of fat cells and a few primitive blood cells. Red marrow is a hematopoietic tissue and is the site of production of erythrocytes and granular leukocytes. Bone marrow is made up of a framework of connective tissue containing branching fibers with the frame being filled with marrow cells. Bone Marrow: Composition and Hematopoiesis.
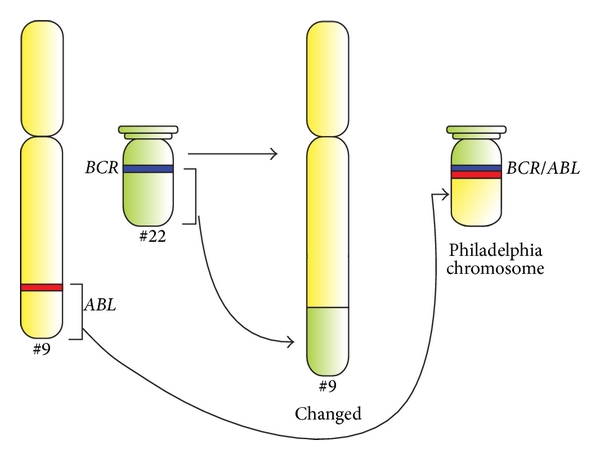
Schematic diagram of the translocation that forms the Philadelphia chromosome, and schematic representation of the BCR and ABL genes.
ABL1 and BCR genes reside on the long arms of chromosomes 9 and 22, respectively. The fusion BCR-ABL gene is formed with the translocation of the ABL1 gene (from chromosome 9) to the long arm of chromosome 22, creating a changed chromosome 22 (the so-called Philadelphia chromosome).
The symptoms discussed above vary in severity depending on the 3 disease phases of CML (based on the immature WBCs or blast cells in the blood or bone marrow Bone marrow The soft tissue filling the cavities of bones. Bone marrow exists in two types, yellow and red. Yellow marrow is found in the large cavities of large bones and consists mostly of fat cells and a few primitive blood cells. Red marrow is a hematopoietic tissue and is the site of production of erythrocytes and granular leukocytes. Bone marrow is made up of a framework of connective tissue containing branching fibers with the frame being filled with marrow cells. Bone Marrow: Composition and Hematopoiesis).
Chronic stable phase:
Accelerated phase:
Blast crisis (or acute phase Acute phase Short Bowel Syndrome):
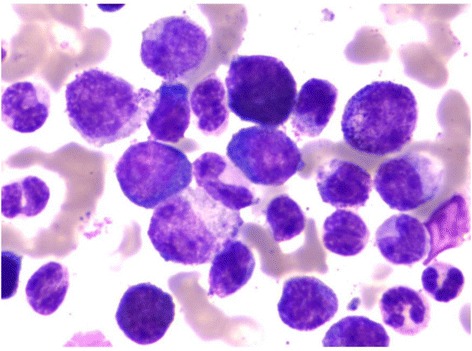
Bone marrow examination:
Blasts seen in the bone marrow of a patient diagnosed with CML
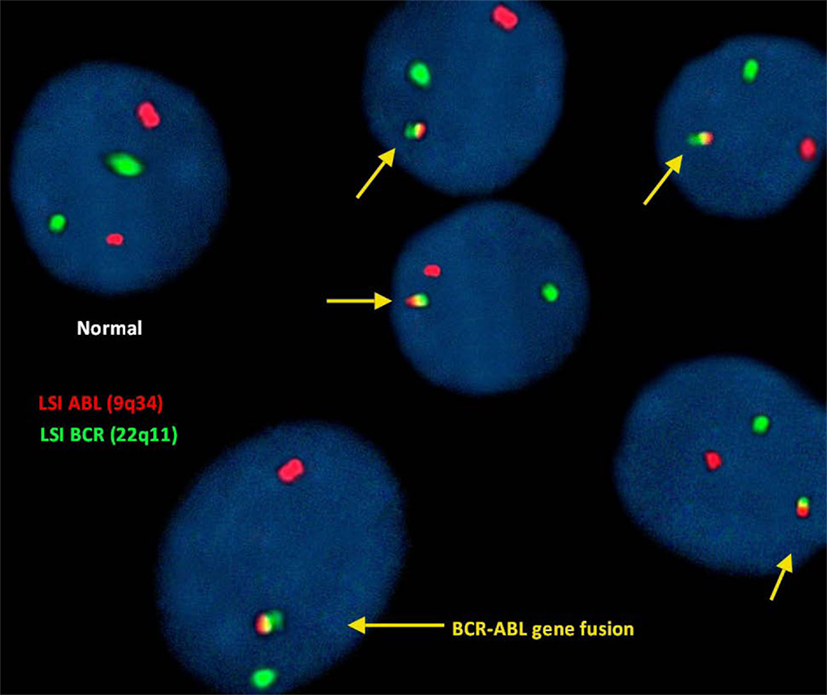
FISH for the detection of (9;22)(q34;q11):
In this study, FISH analysis of the ABL (9q34) gene was identified as fluorescent red dots and the BCR (22q11) gene as green dots. Therefore, a cell exhibiting 2 separate green and red dots was considered a normal cell showing no translocation.
However, the irregular translocation in a cell was identified by 1 red and 1 green and fused red, yellow, and green signals (representing BCR-ABL gene fusion).
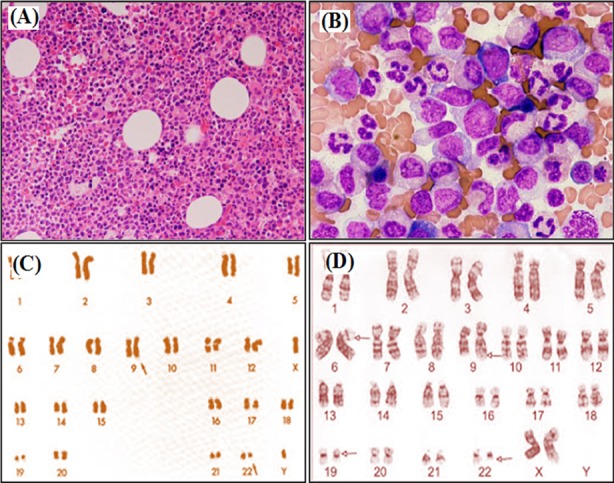
Bone marrow studies and karyotyping of CML:
Bone marrow aspirates showing hypercellularity
A: x100 magnification
B: x400 magnification
C: standard Philadelphia chromosome (altered chromosome 22) karyotype analysis
D: CML with more complex involvement of chromosome 6
Myeloproliferative neoplasms Neoplasms New abnormal growth of tissue. Malignant neoplasms show a greater degree of anaplasia and have the properties of invasion and metastasis, compared to benign neoplasms. Benign Bone Tumors can be compared with the following WHO classification:
| Disease | Mutations | Key points |
|---|---|---|
| CML | BCR-ABL1 ( Philadelphia chromosome Philadelphia chromosome An aberrant form of human chromosome 22 characterized by translocation of the distal end of chromosome 9 from 9q34, to the long arm of chromosome 22 at 22q11. It is present in the bone marrow cells of 80 to 90 percent of patients with chronic myelocytic leukemia. Acute Lymphoblastic Leukemia) | Proliferation of mature and maturing granulocytes Granulocytes Leukocytes with abundant granules in the cytoplasm. They are divided into three groups according to the staining properties of the granules: neutrophilic, eosinophilic, and basophilic. Mature granulocytes are the neutrophils; eosinophils; and basophils. White Myeloid Cells: Histology |
| Essential thrombocythemia Essential thrombocythemia Essential thrombocythemia (ET) is a type of myeloproliferative neoplasm characterized by the clonal thrombocytosis linked to somatic mutations involving Janus kinase 2 (JAK2), calreticulin (CALR), and myeloproliferative leukemia virus oncogene (MPL). Patients can be asymptomatic or present with vasomotor symptoms. Essential Thrombocythemia | JAK2, CALR, or MPL | Excessive clonal platelet production |
| Polycythemia Polycythemia An increase in the total red cell mass of the blood. Renal Cell Carcinoma vera | JAK2 | Elevated RBC mass Mass Three-dimensional lesion that occupies a space within the breast Imaging of the Breast |
| Primary myelofibrosis Primary myelofibrosis Primary myelofibrosis (PMF) is a myeloproliferative neoplasm characterized by chronic myeloproliferation with nonclonal fibroblastic deposition, resulting in bone marrow fibrosis. The abnormality stems from genetic mutations of the hematopoietic stem cells (typically, JAK2 mutation). Primary symptoms are anemia and extramedullary hematopoiesis,. Primary Myelofibrosis | JAK2, CALR, or MPL | Obliterative bone marrow fibrosis Bone Marrow Fibrosis Primary Myelofibrosis |
Other types:
Chronic stable phase:
Accelerated phase:
Blast crisis: