


A leucemia mielóide crónica (LMC) é uma proliferação maligna da linhagem celular granulocítica, caracterizada por uma diferenciação relativamente normal. A anomalia genética subjacente é o cromossoma Filadélfia, uma alteração do cromossoma 22, que resulta da translocação recíproca (9;22)(q34; q11). O cromossoma contém o gene Gene A category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms. Basic Terms of Genetics de fusão BCR-ABL1 (ABL1 no cromossoma 9 e BCR BCR Lymphocytes: Histology no cromossoma 22), que induz a ativação constitutiva da tirosina quinase e, consequentemente, a produção descontrolada de granulócitos. Os doentes com LMC podem ser assintomáticos ou apresentar sintomas constitucionais, dor esternal e esplenomegalia. As análises laboratoriais revelam aumento de leucócitos e o esfregaço de sangue periférico mostra um aumento do número de células imaturas. A demonstração da translocação Filadélfia através de técnicas citogenéticas é considerada o melhor teste diagnóstico (gold standard). Sem tratamento, a LMC apresenta geralmente um curso trifásico, progredindo de uma fase crónica para uma fase acelerada e, por fim, uma crise blástica terminal. O tratamento inclui terapêutica com inibidores da tirosina quinase e agentes paliativos, bem como transplante de células hematopoéticas alogénico.
Last updated: Mar 28, 2022
A leucemia mielóide crónica (LMC) é uma neoplasia mieloproliferativa crónica caracterizada pela proliferação descontrolada e desregulada da linhagem granulocítica (células maduras e em maturação), com capacidade de diferenciação mantida. A leucemia mielóide crónica também é conhecida como leucemia mielocítica crónica ou leucemia mielógena crónica.
A hematopoiese inicia-se com uma célula estaminal hematopoiética, estimulada a dividir-se e diferenciar-se com estímulos químicos apropriados (fatores de crescimento hemopoiéticos).
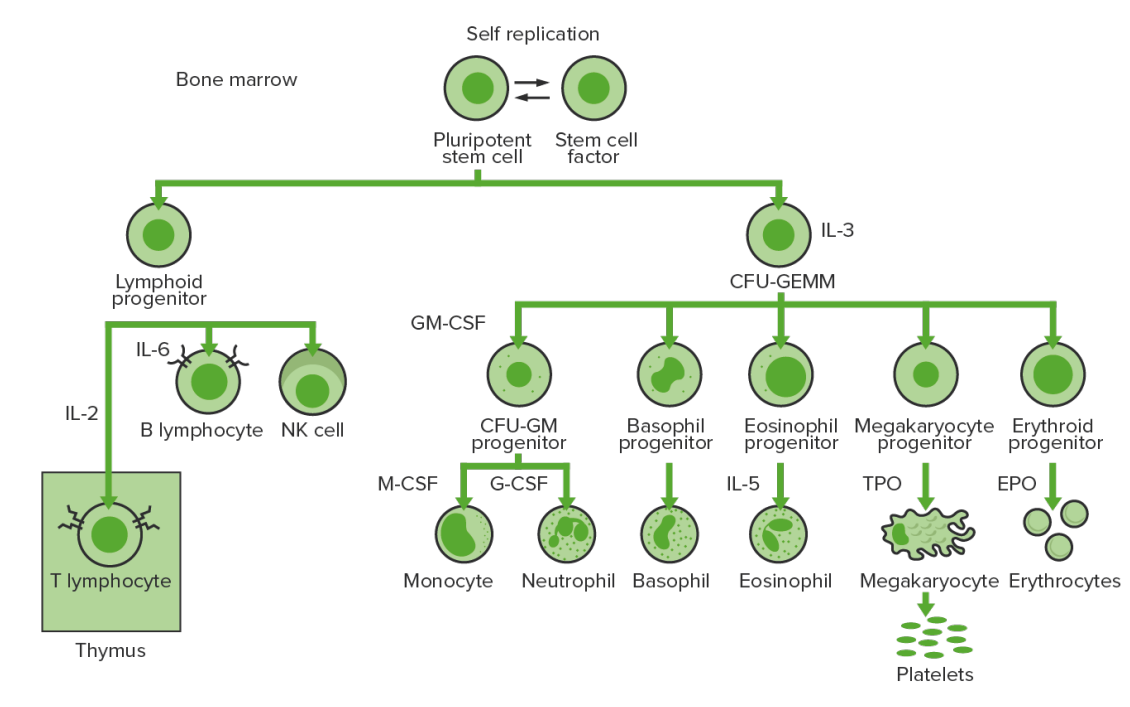
Hematopoiese da medula óssea:
Proliferação e diferenciação dos elementos sanguíneos:
Na LMC, ocorre proliferação sustentada de células da linhagem granulocítica (mieloblastos → neutrófilos, basófilos, eosinófilos). É possível observar células maduras e células em maturação; assim, existem células que são apenas parcialmente eficazes.
CFU-GEMM (pela sigla em inglês): unidade formadora de colónias de granulócitos, eritrócitos, monócitos, megacariócitos
GM-CSF (pela sigla em inglês): fator estimulador de colónias de macrófagos e granulócitos
M-CSF (pela sigla em inglês): fator estimulador de colónias de macrófagos
G-CSF (pela sigla em inglês): fator estimulador de colónias de granulócitos
NK: natural killer
A leucemia mielóide crónica é uma anomalia adquirida das células estaminais hematopoiéticas na medula óssea.
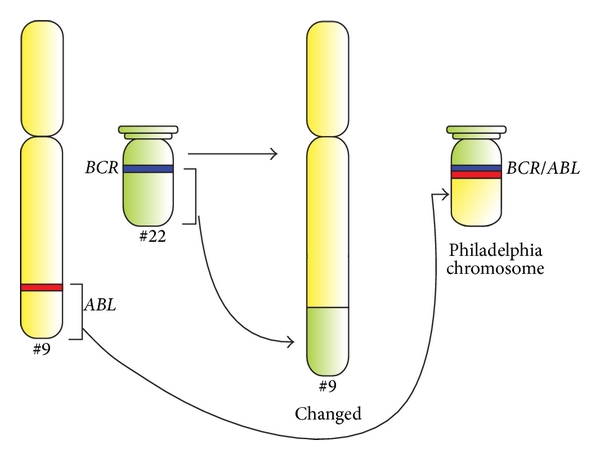
Diagrama esquemático da translocação genética característica do cromossoma Filadélfia e representação esquemática dos genes BCR e ABL.
Os genes ABL1 e BCR pertencem aos braços longos dos cromossomas 9 e 22, respetivamente. O gene de fusão BCR-ABL é formado pela translocação do gene ABL1 (do cromossoma 9) para o braço longo do cromossoma 22, criando um cromossoma 22 alterado (o chamado cromossoma Filadélfia).
Os sintomas acima descritos variam em gravidade, dependendo das 3 fases de doença da LMC (com base em leucócitos imaturos ou blastos no sangue ou medula óssea).
Fase crónica estável:
Fase acelerada:
Crise blástica (ou fase aguda):
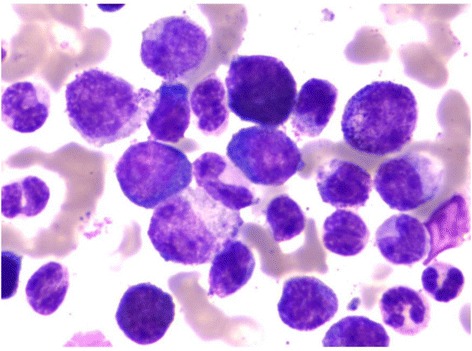
Avaliação da medula óssea:
Observam-se blastos na medula óssea de um doente com diagnóstico de LMC
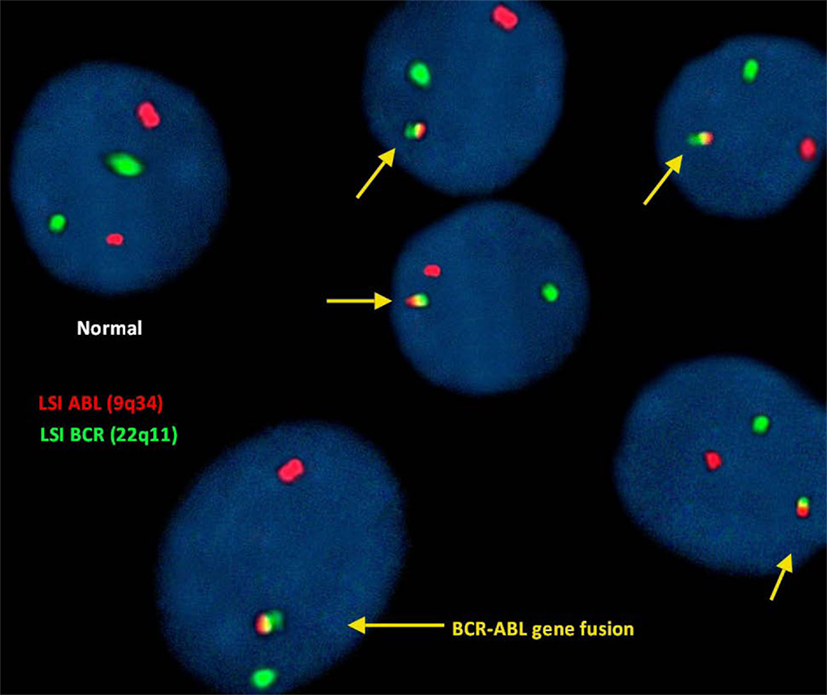
FISH para a deteção de t(9;22)(q34; q11):
Neste estudo, a análise FISH do gene ABL (9q34) foi identificada com pontos vermelhos fluorescentes e o gene BCR (22q11) com pontos verdes. Portanto, uma célula que exiba 2 pontos verdes e vermelhos separados foi considerada uma célula normal que não revela translocação.
No entanto, a translocação irregular numa célula é identificada com 1 ponto vermelho e 1 ponto verde e sinais vermelhos, amarelos e verdes fundidos (representativo da fusão do gene BCR-ABL).
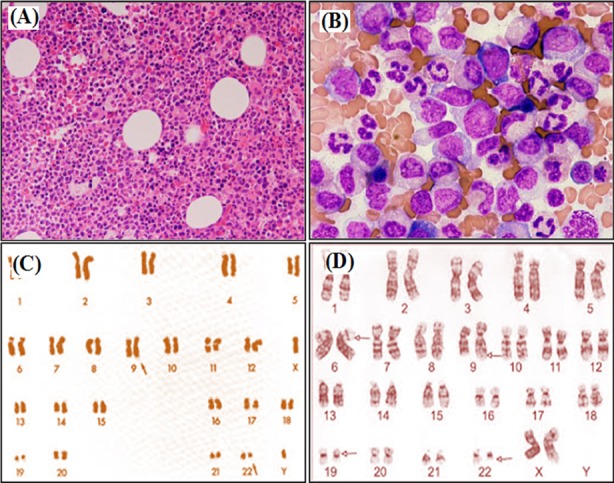
Avaliação de medula óssea e cariótipo de LMC:
Aspirado de medula óssea revela hipercelularidade
A: ampliação x100
B: ampliação x400
C: análise padrão do cariótipo do cromossoma Filadélfia (cromossoma 22 alterado)
D: LMC com envolvimento mais complexo do cromossoma 6
As neoplasias mieloproliferativas podem ser comparadas com a seguinte classificação da Organização Mundial da Saúde (OMS):
| Doença | Mutações | Pontos chave |
|---|---|---|
| LMC | BCR-ABL1 (cromossoma Filadélfia) | Proliferação de granulócitos maduros e em maturação |
| Trombocitemia essencial | JAK2, CALR ou MPL | Produção clonal excessiva de plaquetas |
| Policitemia vera | JAK2 | Aumento da massa eritróide |
| Mielofibrose primária | JAK2, CALR ou MPL | Fibrose obliterativa da medula óssea |
Outros tipos:
Fase crónica estável:
Fase acelerada:
Crise blástica:
