


A trombocitemia essencial (TE) é um tipo de neoplasia mieloproliferativa caracterizada por trombocitose clonal ligada a mutações somáticas que envolvem a Janus quinase 2 (JAK2), a calreticulina (CALR) e o oncogene do vírus da leucemia mieloproliferativa (MPL). Os doentes podem ser assintomáticos ou apresentar sintomas vasomotores, como cefaleias, eritromelalgia e distúrbios visuais transitórios. O curso clínico pode ser complicado por eventos trombo-hemorrágicos, bem como pela progressão para mielofibrose e LMA. O diagnóstico é baseado em achados laboratoriais de trombocitose, biópsia de medula óssea e estudos genéticos. O tratamento visa reduzir a contagem de plaquetas usando agentes citorredutores (hidroxiureia) e diminuir os episódios de trombose com aspirina e anticoagulação sistémica com base na estratificação do risco de trombose.
Last updated: Jun 30, 2026
A trombocitemia essencial (TE) é uma neoplasia mieloproliferativa crónica caracterizada pela produção excessiva de plaquetas e um aumento da tendência trombótica e hemorrágica. Outros nomes são trombocitose essencial e trombocitose primária.
A hematopoiese começa com uma célula estaminal hematopoiética, que é estimulada a dividir-se e diferenciar por estímulos químicos apropriados (fatores de crescimento hematopoiético).
Produção de plaquetas:
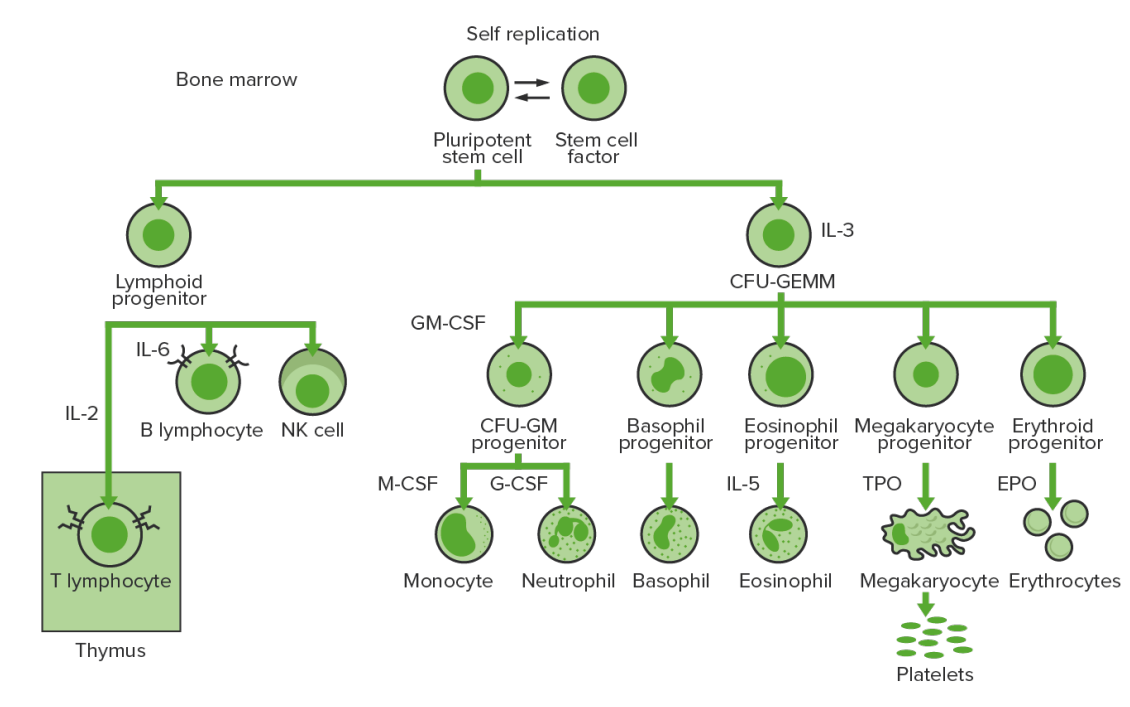
Hematopoiese na medula óssea: proliferação e diferenciação dos elementos sanguíneos.
CFU-GEMM, pela sigla em inglês: unidade formadora de colónias de granulócitos, eritrócitos, monócitos, megacariócitos
CFU-GM, pela sigla em inglês: unidade formadora de colónias de granulócitos-macrófagos
GM-CSF, pela sigla em inglês: fator estimulador de colónias de granulócitos-macrófagos
M-CSF, pela sigla em inglês: fator estimulador de colónias de macrófagos
G-CSF, pela sigla em inglês: fator estimulador de colónias de granulócitos
NK: “natural killer”
TPO: trombopoietina
Aumento da incidência devido a:
O aumento da diátese hemorrágica na TE é devido a:
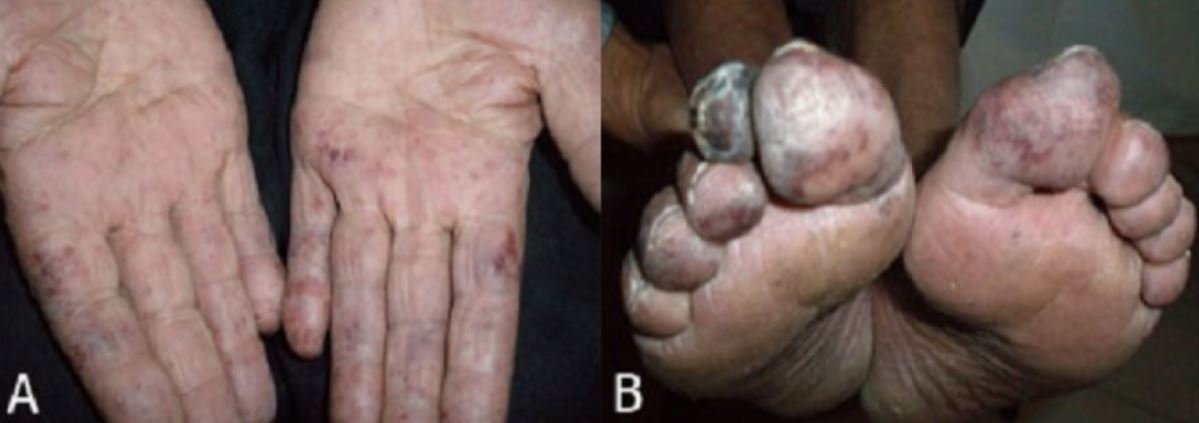
Achados físicos da trombocitemia essencial:
Livedo reticular, áreas de isquemia azulada e mosqueada dos dedos (A) e gangrena do dedo do pé (B)
O diagnóstico requer todos os 4 critérios major ou os 3 primeiros critérios major e 1 critério minor.
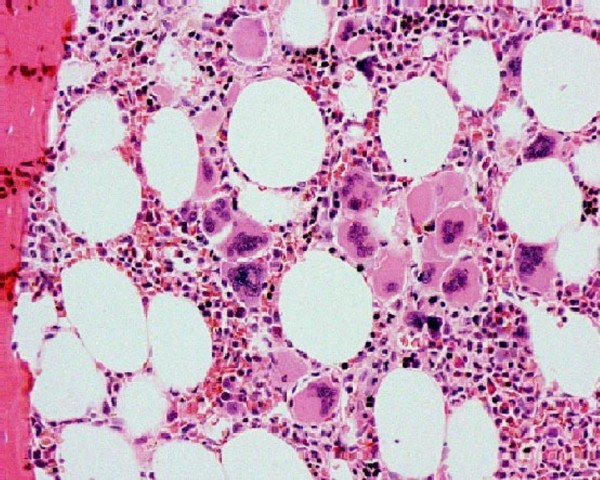
Biópsia da medula óssea na trombocitemia essencial:
Podem ser observados megacariócitos aumentados.
As neoplasias mieloproliferativas podem ser comparadas com a seguinte classificação da OMS:
| Doença | Mutações | Pontos chave |
|---|---|---|
| LMC | BCR-ABL1 (cromossoma Filadélfia) | Proliferação de granulócitos maduros e em maturação |
| TE | JAK2, CALR ou MPL | Produção clonal excessiva de plaquetas |
| Policitemia vera ( PV PV Polycythemia vera (PV) is a chronic myeloproliferative neoplasm characterized by the overproduction of rbcs. In addition, the wbc and platelet counts are also increased, which differentiate pv from erythrocytosis seen with chronic hypoxia and other chronic conditions. Polycythemia Vera) | JAK2 | Aumento da massa de hemácias |
| Mielofibrose primária (MFP) | JAK2, CALR ou MPL | Fibrose obliteradora da medula óssea |
Outros tipos:
| Risco de trombose | Características | Tratamento |
|---|---|---|
| Risco muito baixo |
|
|
| Baixo risco |
|
|
| Risco intermediário |
|
|
| Risco alto |
|
|
