


A mielofibrose primária (MFP) é uma neoplasia mieloproliferativa caracterizada por mieloproliferação crónica com deposição fibroblástica não clonal, resultando em fibrose da medula óssea. A alteração decorre de mutações genéticas das células estaminais hematopoiéticas (tipicamente, mutação JAK2). Os sintomas primários são a anemia Anemia Anemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types e a hematopoiese extramedular, que incluem fadiga severa, perda de peso e hepatoesplenomegalia. Os achados laboratoriais incluem anemia Anemia Anemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types (e outras citopenias) e a biópsia da medula óssea revela fibrose extensa. O tratamento é realizado através de transplante de células estaminais hematopoiéticas e intervenções direcionadas aos sintomas.
Last updated: Jan 7, 2026
A mielofibrose primária (MFP) é uma neoplasia mieloproliferativa crónica caracterizada pela proliferação de células mieloides, com proliferação e hiperatividade de fibroblastos não clonais, resultando em fibrose medular obliterativa. Outros nomes incluem mielofibrose idiopática crónica e metaplasia Metaplasia A condition in which there is a change of one adult cell type to another similar adult cell type. Cellular Adaptation mieloide agnogénica.
A hematopoiese começa com a célula estaminal hematopoiética, que é estimulada a dividir-se e diferenciar-se com estímulos químicos apropriados (fatores de crescimento hematopoiéticos)
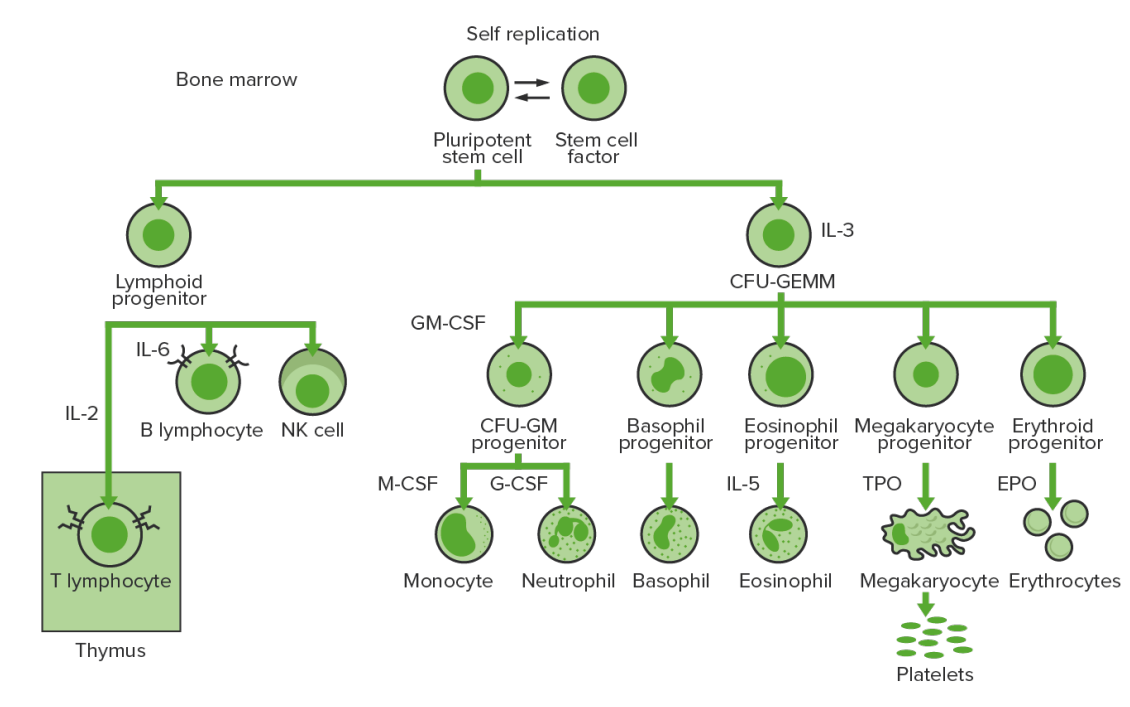
Hematopoiese na medula óssea: proliferação e diferenciação dos elementos figurados sanguíneos.
CFU-GEMM, pela sigla em inglês: unidade formadora de colónias de granulócitos, eritrócitos, monócitos, megacariócitos
CFU-GM, pela sigla em inglês: unidade formadora de colónias de granulócitos-macrófagos
GM-CSF, pela sigla em inglês: fator estimulador de colónias de granulócitos-macrófagos
M-CSF, pela sigla em inglês: fator estimulador de colónias de macrófagos
G-CSF, pela sigla em inglês: fator estimulador de colónias de granulócitos
NK: “natural killer”
TPO: trombopoietina
Mutações em genes Genes A category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms. DNA Types and Structure associados à hematopoiese são observadas numa proporção significativa de casos de MFP.
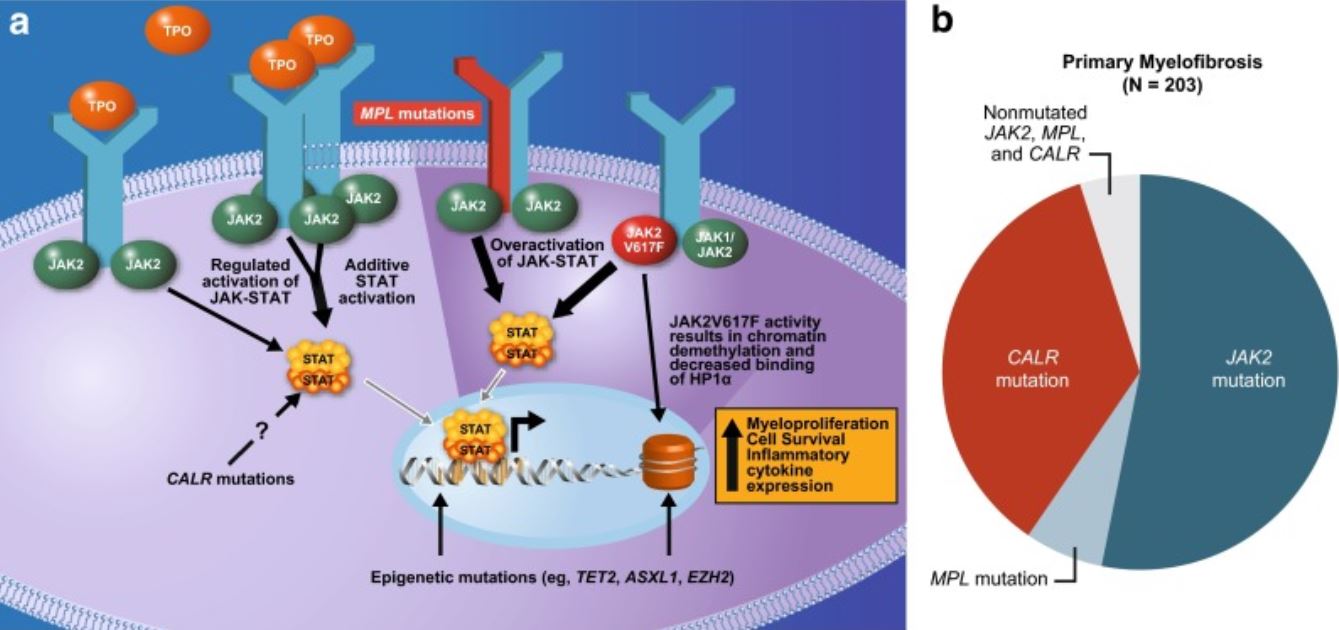
Mutações subjacentes à fisiopatologia da mielofibrose:
Frequentemente afeta a via JAK-STAT
JAK2 (gene JAK2 envolvido): tirosina cinase não recetora que facilita o alcance de sinais extracelulares ao núcleo e a ativação de genes.
MPL ou recetor de trombopoietina (gene MPL envolvido): ativação do recetor de trombopoietina
A calreticulina (gene CALR envolvido) é um regulador da STAT e a mutação causa a ativação do recetor de trombopoietina.
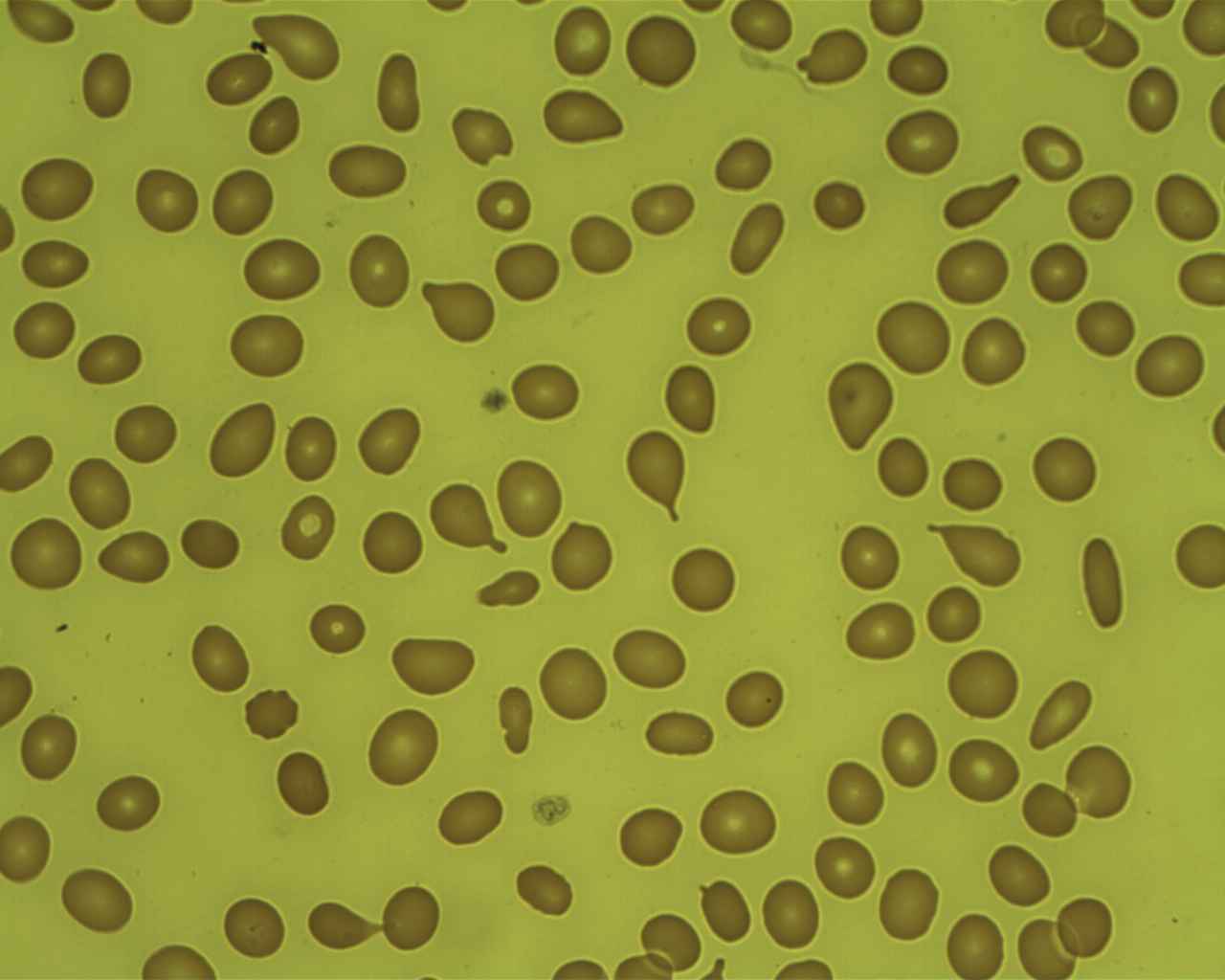
Mielofibrose: células em lágrima em esfregaço de sangue periférico
Imagem: “Tear Drop Poikilocytes” por Prof. Osaro Erhabor. Licença: CC0 1.0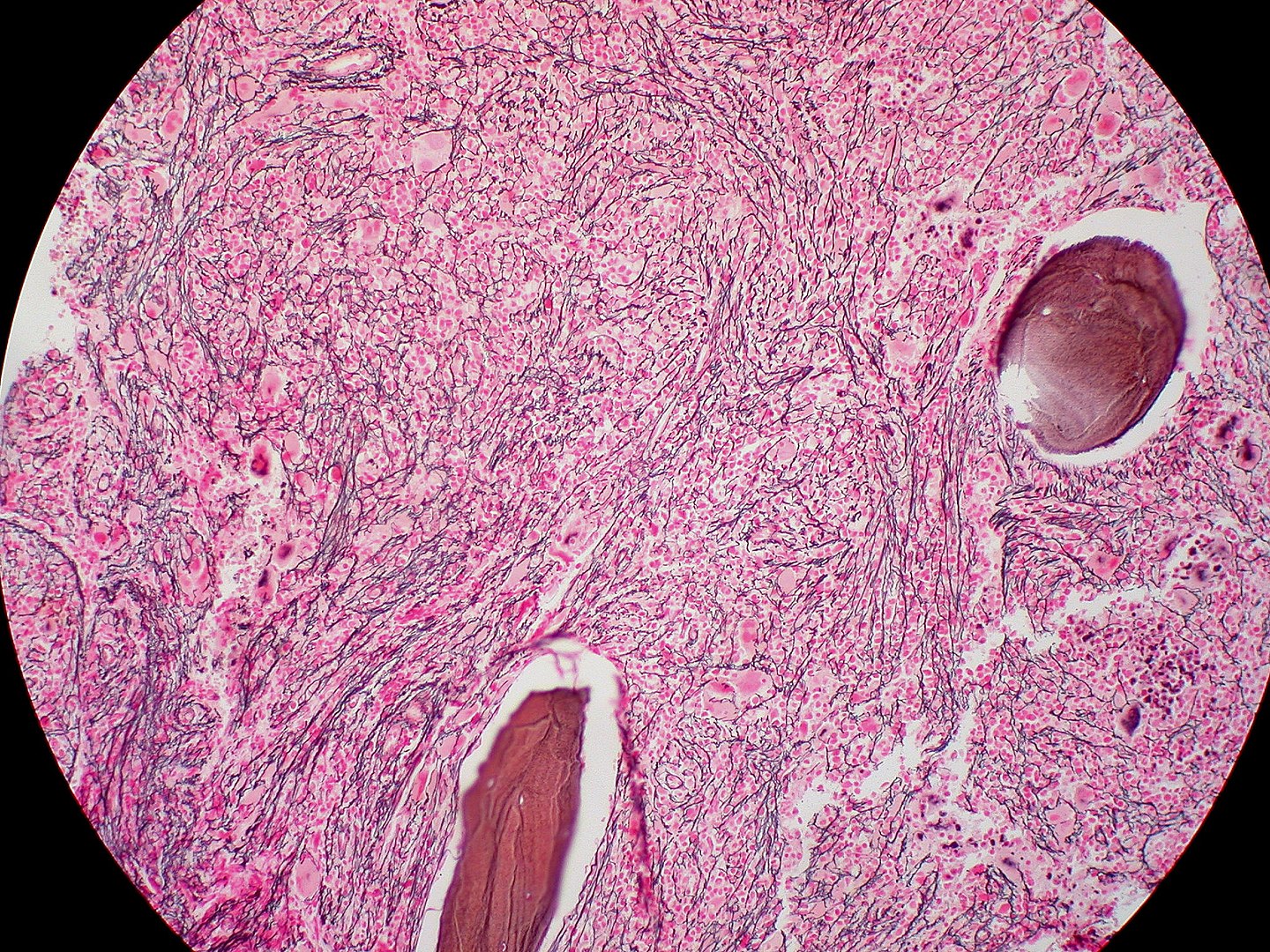
Biópsia da medula óssea na mielofibrose: fibrose extensa (coloração de reticulina)
Imagem: “Myelofibrosis, Reticulin Stain (6032644716)” por Ed Uthman. Licença: CC BY 2.0Em resumo, para lembrar a MFP: Bone Bone Bone is a compact type of hardened connective tissue composed of bone cells, membranes, an extracellular mineralized matrix, and central bone marrow. The 2 primary types of bone are compact and spongy. Bones: Structure and Types marrow is crying ( teardrop cells Teardrop Cells Primary Myelofibrosis) because it is fibrosed and is a dry tap Dry Tap Hairy Cell Leukemia (A medula óssea chora (células em forma de lágrima) porque está fibrosada e é uma torneira seca).
Pré-fibrose/fibrose medular primária precoce (pré-MFP)
Mielofibrose primária fibrótica
As neoplasias mieloproliferativas podem ser comparadas de acordo com a seguinte classificação da OMS:
| Doença | Mutação | Pontos chave |
|---|---|---|
| LMC | BCR-ABL1 (cromossoma Filadelfia) | Proliferação de granulócitos maduros e em maturação |
| TE | JAK2, CALR ou MPL | Produção clonal excessiva de plaquetas |
| Policitemia vera ( PV PV Polycythemia vera (PV) is a chronic myeloproliferative neoplasm characterized by the overproduction of rbcs. In addition, the wbc and platelet counts are also increased, which differentiate pv from erythrocytosis seen with chronic hypoxia and other chronic conditions. Polycythemia Vera) | JAK2 | Aumento da massa eritrocitária |
| MFP | JAK2, CALR ou MPL | Fibrose obliterante da medula óssea |
Outros tipos:
A avaliação do risco é importante na tomada de decisões de tratamento.
