Hairy cell leukemia (HCL) is a rare, chronic, B-cell leukemia characterized by the accumulation of small mature B lymphocytes B lymphocytes Lymphoid cells concerned with humoral immunity. They are short-lived cells resembling bursa-derived lymphocytes of birds in their production of immunoglobulin upon appropriate stimulation. B cells: Types and Functions that have “hair-like projections” visible on microscopy. The abnormal cells accumulate in the peripheral blood, bone marrow Bone marrow The soft tissue filling the cavities of bones. Bone marrow exists in two types, yellow and red. Yellow marrow is found in the large cavities of large bones and consists mostly of fat cells and a few primitive blood cells. Red marrow is a hematopoietic tissue and is the site of production of erythrocytes and granular leukocytes. Bone marrow is made up of a framework of connective tissue containing branching fibers with the frame being filled with marrow cells. Bone Marrow: Composition and Hematopoiesis (causing fibrosis Fibrosis Any pathological condition where fibrous connective tissue invades any organ, usually as a consequence of inflammation or other injury. Bronchiolitis Obliterans), and red pulp Red pulp Secondary Lymphatic Organs of the spleen Spleen The spleen is the largest lymphoid organ in the body, located in the LUQ of the abdomen, superior to the left kidney and posterior to the stomach at the level of the 9th-11th ribs just below the diaphragm. The spleen is highly vascular and acts as an important blood filter, cleansing the blood of pathogens and damaged erythrocytes. Spleen: Anatomy, leading to cytopenias Cytopenias IPEX Syndrome, including anemia Anemia Anemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types, thrombocytopenia Thrombocytopenia Thrombocytopenia occurs when the platelet count is < 150,000 per microliter. The normal range for platelets is usually 150,000-450,000/µL of whole blood. Thrombocytopenia can be a result of decreased production, increased destruction, or splenic sequestration of platelets. Patients are often asymptomatic until platelet counts are < 50,000/µL. Thrombocytopenia, neutropenia Neutropenia Neutrophils are an important component of the immune system and play a significant role in the eradication of infections. Low numbers of circulating neutrophils, referred to as neutropenia, predispose the body to recurrent infections or sepsis, though patients can also be asymptomatic. Neutropenia, and monocytopenia. HCL typically affects middle-aged men and presents with weakness, fatigue Fatigue The state of weariness following a period of exertion, mental or physical, characterized by a decreased capacity for work and reduced efficiency to respond to stimuli. Fibromyalgia, opportunistic infections Infections Invasion of the host organism by microorganisms or their toxins or by parasites that can cause pathological conditions or diseases. Chronic Granulomatous Disease, and splenomegaly Splenomegaly Splenomegaly is pathologic enlargement of the spleen that is attributable to numerous causes, including infections, hemoglobinopathies, infiltrative processes, and outflow obstruction of the portal vein. Splenomegaly (which is often severe). Hairy cell leukemia is diagnosed by identifying hairy cells on a peripheral blood smear Peripheral Blood Smear Anemia: Overview and Types, characteristic cytopenias Cytopenias IPEX Syndrome on CBC/DIFF, bone marrow Bone marrow The soft tissue filling the cavities of bones. Bone marrow exists in two types, yellow and red. Yellow marrow is found in the large cavities of large bones and consists mostly of fat cells and a few primitive blood cells. Red marrow is a hematopoietic tissue and is the site of production of erythrocytes and granular leukocytes. Bone marrow is made up of a framework of connective tissue containing branching fibers with the frame being filled with marrow cells. Bone Marrow: Composition and Hematopoiesis biopsy Biopsy Removal and pathologic examination of specimens from the living body. Ewing Sarcoma, and immunophenotyping Immunophenotyping Process of classifying cells of the immune system based on structural and functional differences. The process is commonly used to analyze and sort T-lymphocytes into subsets based on CD antigens by the technique of flow cytometry. Non-Hodgkin Lymphomas. Management consists of observation for asymptomatic patients Patients Individuals participating in the health care system for the purpose of receiving therapeutic, diagnostic, or preventive procedures. Clinician–Patient Relationship and purine analogs Purine Analogs Antimetabolite Chemotherapy for symptomatic patients Patients Individuals participating in the health care system for the purpose of receiving therapeutic, diagnostic, or preventive procedures. Clinician–Patient Relationship. Both the disease itself and its treatments can cause immunosuppression, so prevention and prompt treatment of infections Infections Invasion of the host organism by microorganisms or their toxins or by parasites that can cause pathological conditions or diseases. Chronic Granulomatous Disease is also important.
Last updated: Dec 15, 2025
Hairy cell leukemia (HCL) is a rare, chronic B-cell leukemia characterized by the accumulation of small mature B lymphocytes B lymphocytes Lymphoid cells concerned with humoral immunity. They are short-lived cells resembling bursa-derived lymphocytes of birds in their production of immunoglobulin upon appropriate stimulation. B cells: Types and Functions that have hair-like projections visible on microscopy.
Hairy cell leukemia is a clonal B-cell malignancy Malignancy Hemothorax. The exact etiology is unknown, but several environmental exposures appear to increase the risk of developing HCL.
The underlying issue in HCL is that the bone marrow Bone marrow The soft tissue filling the cavities of bones. Bone marrow exists in two types, yellow and red. Yellow marrow is found in the large cavities of large bones and consists mostly of fat cells and a few primitive blood cells. Red marrow is a hematopoietic tissue and is the site of production of erythrocytes and granular leukocytes. Bone marrow is made up of a framework of connective tissue containing branching fibers with the frame being filled with marrow cells. Bone Marrow: Composition and Hematopoiesis produces far too many B lymphocytes B lymphocytes Lymphoid cells concerned with humoral immunity. They are short-lived cells resembling bursa-derived lymphocytes of birds in their production of immunoglobulin upon appropriate stimulation. B cells: Types and Functions, which crowd out the other types of blood cells. These abnormal B cells B cells Lymphoid cells concerned with humoral immunity. They are short-lived cells resembling bursa-derived lymphocytes of birds in their production of immunoglobulin upon appropriate stimulation. B cells: Types and Functions are unable to protect the body from infectious invaders the way normal B cells B cells Lymphoid cells concerned with humoral immunity. They are short-lived cells resembling bursa-derived lymphocytes of birds in their production of immunoglobulin upon appropriate stimulation. B cells: Types and Functions can.
The pathogenesis is incompletely understood, but most cases appear to arise from late activated memory Memory Complex mental function having four distinct phases: (1) memorizing or learning, (2) retention, (3) recall, and (4) recognition. Clinically, it is usually subdivided into immediate, recent, and remote memory. Psychiatric Assessment B cells B cells Lymphoid cells concerned with humoral immunity. They are short-lived cells resembling bursa-derived lymphocytes of birds in their production of immunoglobulin upon appropriate stimulation. B cells: Types and Functions, which acquire an activating BRAF V600E gene mutation Gene Mutation Myotonic Dystrophies.
The diagnosis is based on identifying hairy cells on a peripheral blood smear Peripheral Blood Smear Anemia: Overview and Types, immunohistochemistry Immunohistochemistry Histochemical localization of immunoreactive substances using labeled antibodies as reagents. Myeloperoxidase Deficiency, and/or flow cytometry Flow cytometry Technique using an instrument system for making, processing, and displaying one or more measurements on individual cells obtained from a cell suspension. Cells are usually stained with one or more fluorescent dyes specific to cell components of interest, e.g., DNA, and fluorescence of each cell is measured as it rapidly transverses the excitation beam (laser or mercury arc lamp). Fluorescence provides a quantitative measure of various biochemical and biophysical properties of the cell, as well as a basis for cell sorting. Other measurable optical parameters include light absorption and light scattering, the latter being applicable to the measurement of cell size, shape, density, granularity, and stain uptake. X-linked Agammaglobulinemia and, potentially, a bone marrow Bone marrow The soft tissue filling the cavities of bones. Bone marrow exists in two types, yellow and red. Yellow marrow is found in the large cavities of large bones and consists mostly of fat cells and a few primitive blood cells. Red marrow is a hematopoietic tissue and is the site of production of erythrocytes and granular leukocytes. Bone marrow is made up of a framework of connective tissue containing branching fibers with the frame being filled with marrow cells. Bone Marrow: Composition and Hematopoiesis biopsy Biopsy Removal and pathologic examination of specimens from the living body. Ewing Sarcoma.
Laboratory findings may include:
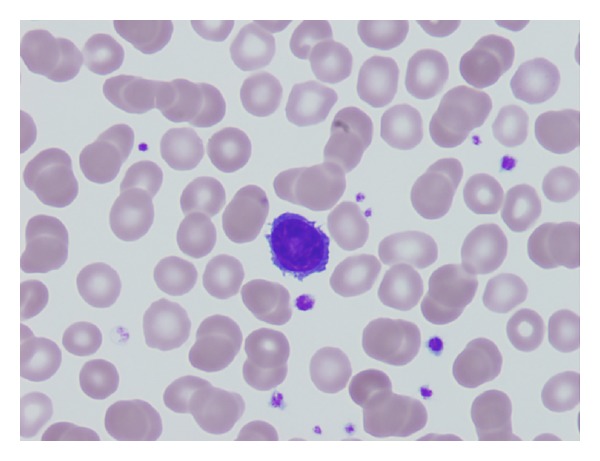
Peripheral blood smear showing a hairy cell
Image: “Primary hairy cell leukemia/lymphoma of the breast: a case report and review of the literature” by Pilichowska M, Shariftabrizi A, Mukand-Cerro I, Miller K. License: CC BY 3.0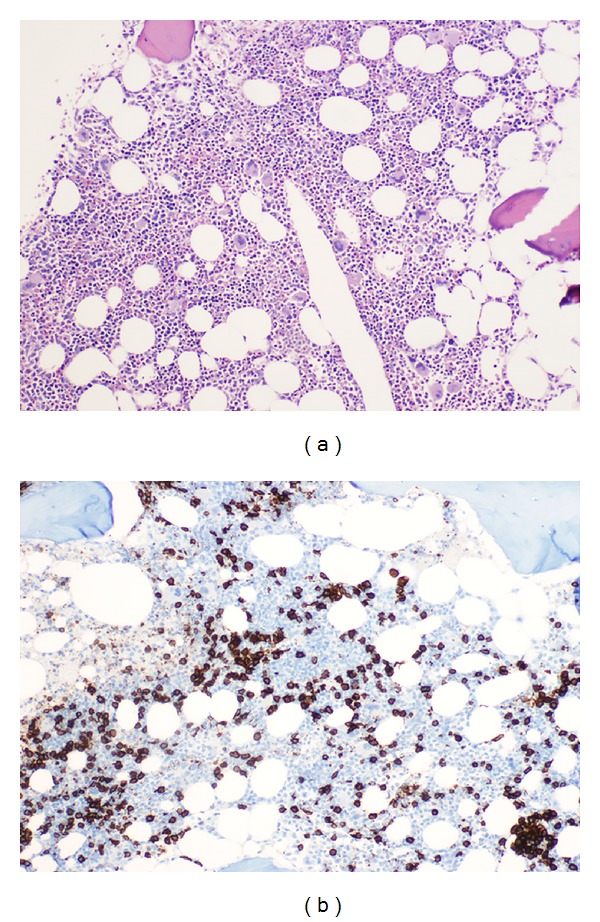
Bone marrow biopsy:
(a): Rather inconspicuous diffuse infiltrate of small mature lymphocytes (H&E, ×200).
(b): Immunohistochemical staining for CD20 reveals B-cell nature of the infiltrate (immunoperoxidase, ×200).
Asymptomatic patients Patients Individuals participating in the health care system for the purpose of receiving therapeutic, diagnostic, or preventive procedures. Clinician–Patient Relationship should be observed rather than treated.
Treatment is indicated in the following clinical situations: