Paroxysmal nocturnal hemoglobinuriaHemoglobinuriaThe presence of free hemoglobin in the urine, indicating hemolysis of erythrocytes within the vascular system. After saturating the hemoglobin-binding proteins (haptoglobins), free hemoglobin begins to appear in the urine.Transfusion Reactions (PNH) is a rare but serious acquired hemolytic anemiaHemolytic AnemiaHemolytic anemia (HA) is the term given to a large group of anemias that are caused by the premature destruction/hemolysis of circulating red blood cells (RBCs). Hemolysis can occur within (intravascular hemolysis) or outside the blood vessels (extravascular hemolysis). Hemolytic Anemia with periodic exacerbations. This anemiaAnemiaAnemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types is caused by nonmalignant clonal expansionClonal ExpansionSeborrheic Keratosis of ≥ 1 hematopoietic stem cellsHematopoietic stem cellsProgenitor cells from which all blood cells derived. They are found primarily in the bone marrow and also in small numbers in the peripheral blood.Bone Marrow: Composition and Hematopoiesis that have acquired a somatic mutationSomatic MutationNon-Hodgkin Lymphomas of the phosphatidylinositol N-acetylglucosaminyltransferase subunit A (PIG-A) geneGeneA category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms.Basic Terms of Genetics. Clonal expansionClonal ExpansionSeborrheic Keratosis of affected stem cells are deficient in glycosylphosphatidylinositol-anchored proteinsProteinsLinear polypeptides that are synthesized on ribosomes and may be further modified, crosslinked, cleaved, or assembled into complex proteins with several subunits. The specific sequence of amino acids determines the shape the polypeptide will take, during protein folding, and the function of the protein.Energy Homeostasis (GPI-APs). Deficiency of the GPI-APs CD55 and CD59, which regulate complement, results in intravascular hemolysisIntravascular hemolysisHemolytic Anemia. The classic triad of PNH is hemolytic anemiaHemolytic AnemiaHemolytic anemia (HA) is the term given to a large group of anemias that are caused by the premature destruction/hemolysis of circulating red blood cells (RBCs). Hemolysis can occur within (intravascular hemolysis) or outside the blood vessels (extravascular hemolysis). Hemolytic Anemia, marrow failure, and thrombophiliaThrombophiliaA disorder of hemostasis in which there is a tendency for the occurrence of thrombosis.Hypercoagulable States. PatientsPatientsIndividuals participating in the health care system for the purpose of receiving therapeutic, diagnostic, or preventive procedures.Clinician–Patient Relationship may be treated with the monoclonal antibody eculizumab or with stem cell transplantation, in addition to being treated for associated complications.
PrevalencePrevalenceThe total number of cases of a given disease in a specified population at a designated time. It is differentiated from incidence, which refers to the number of new cases in the population at a given time.Measures of Disease Frequency: approximately 5 cases per million patientsPatientsIndividuals participating in the health care system for the purpose of receiving therapeutic, diagnostic, or preventive procedures.Clinician–Patient Relationship
SexSexThe totality of characteristics of reproductive structure, functions, phenotype, and genotype, differentiating the male from the female organism.Gender Dysphoria ratio: men = women
Ages affected: mostly young adults; also children and those > 70 years
Etiology
X-linkedX-linkedGenetic diseases that are linked to gene mutations on the X chromosome in humans or the X chromosome in other species. Included here are animal models of human X-linked diseases.Common Variable Immunodeficiency (CVID)somatic mutationSomatic MutationNon-Hodgkin Lymphomas of the phosphatidylinositol N-acetylglucosaminyltransferase subunit A (PIG-A) geneGeneA category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms.Basic Terms of Genetics in a multipotent hematopoietic stem cell
PIG-AgeneGeneA category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms.Basic Terms of Genetics: responsible for the 1st step in synthesisSynthesisPolymerase Chain Reaction (PCR) of glycosylphosphatidylinositol (GPI)
GPI anchors dozens of proteinsProteinsLinear polypeptides that are synthesized on ribosomes and may be further modified, crosslinked, cleaved, or assembled into complex proteins with several subunits. The specific sequence of amino acids determines the shape the polypeptide will take, during protein folding, and the function of the protein.Energy Homeostasis to the cell surface, including complement inhibitors (regulators) CD59 and CD55.
CD59: protects RBCsRBCsErythrocytes, or red blood cells (RBCs), are the most abundant cells in the blood. While erythrocytes in the fetus are initially produced in the yolk sac then the liver, the bone marrow eventually becomes the main site of production.Erythrocytes: Histology against membrane attack complexMembrane attack complexA product of complement activation cascade, regardless of the pathways, that forms transmembrane channels causing disruption of the target cell membrane and cell lysis. It is formed by the sequential assembly of terminal complement components (complement C5b; complement C6; complement C7; complement C8; and complement C9) into the target membrane. The resultant C5b-8-poly-c9 is the ‘membrane attack complex’ or MAC.Type II Hypersensitivity Reaction (MAC)–mediated intravascular hemolysisIntravascular hemolysisHemolytic Anemia
CD55: protects RBCsRBCsErythrocytes, or red blood cells (RBCs), are the most abundant cells in the blood. While erythrocytes in the fetus are initially produced in the yolk sac then the liver, the bone marrow eventually becomes the main site of production.Erythrocytes: Histology against opsonization by complement C3bComplement C3bThe larger fragment generated from the cleavage of complement C3 by C3 convertase. It is a constituent of the alternative pathway C3 convertase (c3bbb), and complement C5 convertases in both the classical (c4b2a3b) and the alternative (c3bbb3b) pathway. C3b participates in immune adherence reaction and enhances phagocytosis. It can be inactivated (ic3b) or cleaved by various proteases to yield fragments such as complement C3c; complement C3d; C3e; C3f; and C3g.C3 Deficiency and subsequent extravascular hemolysisExtravascular hemolysisHemolytic Anemia
RBCsRBCsErythrocytes, or red blood cells (RBCs), are the most abundant cells in the blood. While erythrocytes in the fetus are initially produced in the yolk sac then the liver, the bone marrow eventually becomes the main site of production.Erythrocytes: Histology may show partial or complete absence of GPI-anchored proteinsProteinsLinear polypeptides that are synthesized on ribosomes and may be further modified, crosslinked, cleaved, or assembled into complex proteins with several subunits. The specific sequence of amino acids determines the shape the polypeptide will take, during protein folding, and the function of the protein.Energy Homeostasis (APs).
BenignBenignFibroadenomaclonal expansionClonal ExpansionSeborrheic Keratosis of the mutated stem cells gives rise to CD59/CD55-deficient RBCsRBCsErythrocytes, or red blood cells (RBCs), are the most abundant cells in the blood. While erythrocytes in the fetus are initially produced in the yolk sac then the liver, the bone marrow eventually becomes the main site of production.Erythrocytes: Histology.
Classification
Classic paroxysmal nocturnal hemoglobinuriaHemoglobinuriaThe presence of free hemoglobin in the urine, indicating hemolysis of erythrocytes within the vascular system. After saturating the hemoglobin-binding proteins (haptoglobins), free hemoglobin begins to appear in the urine.Transfusion Reactions (PNH):
No evidence of a preexisting defined bone marrowBone marrowThe soft tissue filling the cavities of bones. Bone marrow exists in two types, yellow and red. Yellow marrow is found in the large cavities of large bones and consists mostly of fat cells and a few primitive blood cells. Red marrow is a hematopoietic tissue and is the site of production of erythrocytes and granular leukocytes. Bone marrow is made up of a framework of connective tissue containing branching fibers with the frame being filled with marrow cells.Bone Marrow: Composition and Hematopoiesis abnormality
PNH in the setting of a specified bone marrowBone marrowThe soft tissue filling the cavities of bones. Bone marrow exists in two types, yellow and red. Yellow marrow is found in the large cavities of large bones and consists mostly of fat cells and a few primitive blood cells. Red marrow is a hematopoietic tissue and is the site of production of erythrocytes and granular leukocytes. Bone marrow is made up of a framework of connective tissue containing branching fibers with the frame being filled with marrow cells.Bone Marrow: Composition and Hematopoiesis disorder:
Clinical and laboratory evidence of hemolysis
Comorbid preexisting defined marrow abnormality (or history of one):
Aplastic anemiaAplastic AnemiaAplastic anemia (AA) is a rare, life-threatening condition characterized by pancytopenia and hypocellularity of the bone marrow (in the absence of any abnormal cells) reflecting damage to hematopoietic stem cells. Aplastic anemia can be acquired or inherited, however, most cases of AA are acquired and caused by autoimmune damage to hematopoietic stem cells. Aplastic Anemia
Myelodysplastic syndrome
Myelofibrosis
Subclinical PNH (PNH-sc):
Comorbid preexisting defined marrow abnormality (or a history of one):
Aplastic anemiaAplastic AnemiaAplastic anemia (AA) is a rare, life-threatening condition characterized by pancytopenia and hypocellularity of the bone marrow (in the absence of any abnormal cells) reflecting damage to hematopoietic stem cells. Aplastic anemia can be acquired or inherited, however, most cases of AA are acquired and caused by autoimmune damage to hematopoietic stem cells. Aplastic Anemia
Myelodysplastic syndrome
Myelofibrosis
Small populations of GPI-AP–deficient erythrocytesErythrocytesErythrocytes, or red blood cells (RBCs), are the most abundant cells in the blood. While erythrocytes in the fetus are initially produced in the yolk sac then the liver, the bone marrow eventually becomes the main site of production. Erythrocytes: Histology and/or granulocytesGranulocytesLeukocytes with abundant granules in the cytoplasm. They are divided into three groups according to the staining properties of the granules: neutrophilic, eosinophilic, and basophilic. Mature granulocytes are the neutrophils; eosinophils; and basophils.White Myeloid Cells: Histology detected by flowFlowBlood flows through the heart, arteries, capillaries, and veins in a closed, continuous circuit. Flow is the movement of volume per unit of time. Flow is affected by the pressure gradient and the resistance fluid encounters between 2 points. Vascular resistance is the opposition to flow, which is caused primarily by blood friction against vessel walls.Vascular Resistance, Flow, and Mean Arterial Pressure cytometric analysis
PIG-A geneGeneA category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms.Basic Terms of Genetics (normal physiology):
Located on the X chromosomeX chromosomeThe female sex chromosome, being the differential sex chromosome carried by half the male gametes and all female gametes in human and other male-heterogametic species.Basic Terms of Genetics
Codes for GPI:
Anchors certain proteinsProteinsLinear polypeptides that are synthesized on ribosomes and may be further modified, crosslinked, cleaved, or assembled into complex proteins with several subunits. The specific sequence of amino acids determines the shape the polypeptide will take, during protein folding, and the function of the protein.Energy Homeostasis to cell membraneCell MembraneA cell membrane (also known as the plasma membrane or plasmalemma) is a biological membrane that separates the cell contents from the outside environment. A cell membrane is composed of a phospholipid bilayer and proteins that function to protect cellular DNA and mediate the exchange of ions and molecules. The Cell: Cell Membrane
Anchors CD55 and CD 59 in multiple blood cell lines:
ErythrocytesErythrocytesErythrocytes, or red blood cells (RBCs), are the most abundant cells in the blood. While erythrocytes in the fetus are initially produced in the yolk sac then the liver, the bone marrow eventually becomes the main site of production. Erythrocytes: Histology
NeutrophilsNeutrophilsGranular leukocytes having a nucleus with three to five lobes connected by slender threads of chromatin, and cytoplasm containing fine inconspicuous granules and stainable by neutral dyes.Innate Immunity: Phagocytes and Antigen Presentation
MonocytesMonocytesLarge, phagocytic mononuclear leukocytes produced in the vertebrate bone marrow and released into the blood; contain a large, oval or somewhat indented nucleus surrounded by voluminous cytoplasm and numerous organelles.Innate Immunity: Phagocytes and Antigen Presentation
PlateletsPlateletsPlatelets are small cell fragments involved in hemostasis. Thrombopoiesis takes place primarily in the bone marrow through a series of cell differentiation and is influenced by several cytokines. Platelets are formed after fragmentation of the megakaryocyte cytoplasm. Platelets: Histology
CD55 and CD59 are complement-regulating surface proteinsProteinsLinear polypeptides that are synthesized on ribosomes and may be further modified, crosslinked, cleaved, or assembled into complex proteins with several subunits. The specific sequence of amino acids determines the shape the polypeptide will take, during protein folding, and the function of the protein.Energy Homeostasis:
Prevent binding of complement to circulating blood cells
CD59 (also called membrane inhibitor of reactive lysis (MIRL)) prevents formation of MAC (prevents intravascular hemolysisIntravascular hemolysisHemolytic Anemia).
CD55 (also called decay accelerating factor) prevents opsonization/complement-mediated blood cell destruction in the spleenSpleenThe spleen is the largest lymphoid organ in the body, located in the LUQ of the abdomen, superior to the left kidney and posterior to the stomach at the level of the 9th-11th ribs just below the diaphragm. The spleen is highly vascular and acts as an important blood filter, cleansing the blood of pathogens and damaged erythrocytes. Spleen: Anatomy (prevents extravascular hemolysisExtravascular hemolysisHemolytic Anemia).
PIG-AgeneGeneA category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms.Basic Terms of Genetics (mutationMutationGenetic mutations are errors in DNA that can cause protein misfolding and dysfunction. There are various types of mutations, including chromosomal, point, frameshift, and expansion mutations. Types of Mutations):
MutationMutationGenetic mutations are errors in DNA that can cause protein misfolding and dysfunction. There are various types of mutations, including chromosomal, point, frameshift, and expansion mutations. Types of Mutations of hematopoietic progenitor cells
ErythrocytesErythrocytesErythrocytes, or red blood cells (RBCs), are the most abundant cells in the blood. While erythrocytes in the fetus are initially produced in the yolk sac then the liver, the bone marrow eventually becomes the main site of production. Erythrocytes: Histology
NeutrophilsNeutrophilsGranular leukocytes having a nucleus with three to five lobes connected by slender threads of chromatin, and cytoplasm containing fine inconspicuous granules and stainable by neutral dyes.Innate Immunity: Phagocytes and Antigen Presentation
MonocytesMonocytesLarge, phagocytic mononuclear leukocytes produced in the vertebrate bone marrow and released into the blood; contain a large, oval or somewhat indented nucleus surrounded by voluminous cytoplasm and numerous organelles.Innate Immunity: Phagocytes and Antigen Presentation
PlateletsPlateletsPlatelets are small cell fragments involved in hemostasis. Thrombopoiesis takes place primarily in the bone marrow through a series of cell differentiation and is influenced by several cytokines. Platelets are formed after fragmentation of the megakaryocyte cytoplasm. Platelets: Histology
Blood cells lack surface proteinsProteinsLinear polypeptides that are synthesized on ribosomes and may be further modified, crosslinked, cleaved, or assembled into complex proteins with several subunits. The specific sequence of amino acids determines the shape the polypeptide will take, during protein folding, and the function of the protein.Energy Homeostasis CD55 and CD59:
Uncontrolled amplification of complement systemComplement systemSerum glycoproteins participating in the host defense mechanism of complement activation that creates the complement membrane attack complex. Included are glycoproteins in the various pathways of complement activation (classical complement pathway; alternative complement pathway; and lectin complement pathway).Innate Immunity: Barriers, Complement, and Cytokines
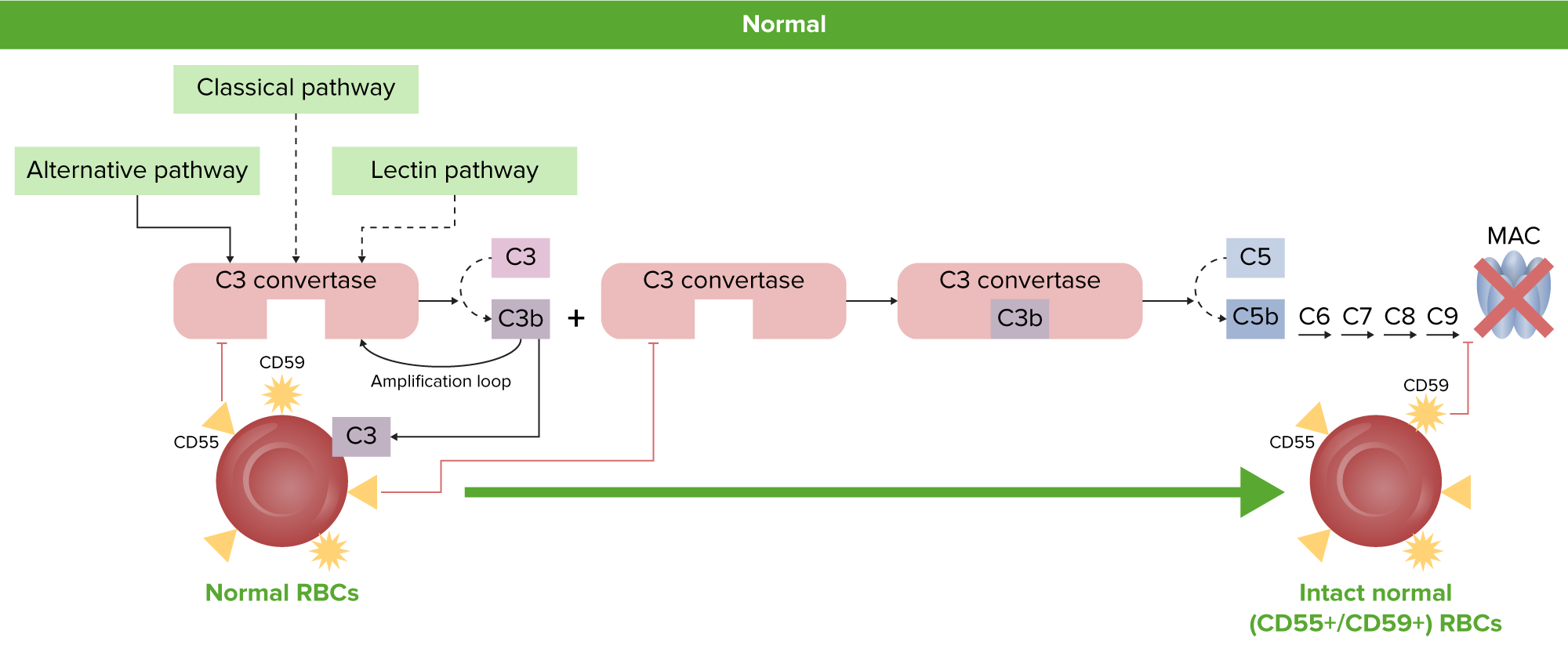
Normal RBC physiology: Normal RBCs are protected from complement activation and hemolysis by the GPI-linked complement regulatory proteins, CD55 and CD59. GPI: glycosylphosphatidylinositol MAC: membrane attack complex
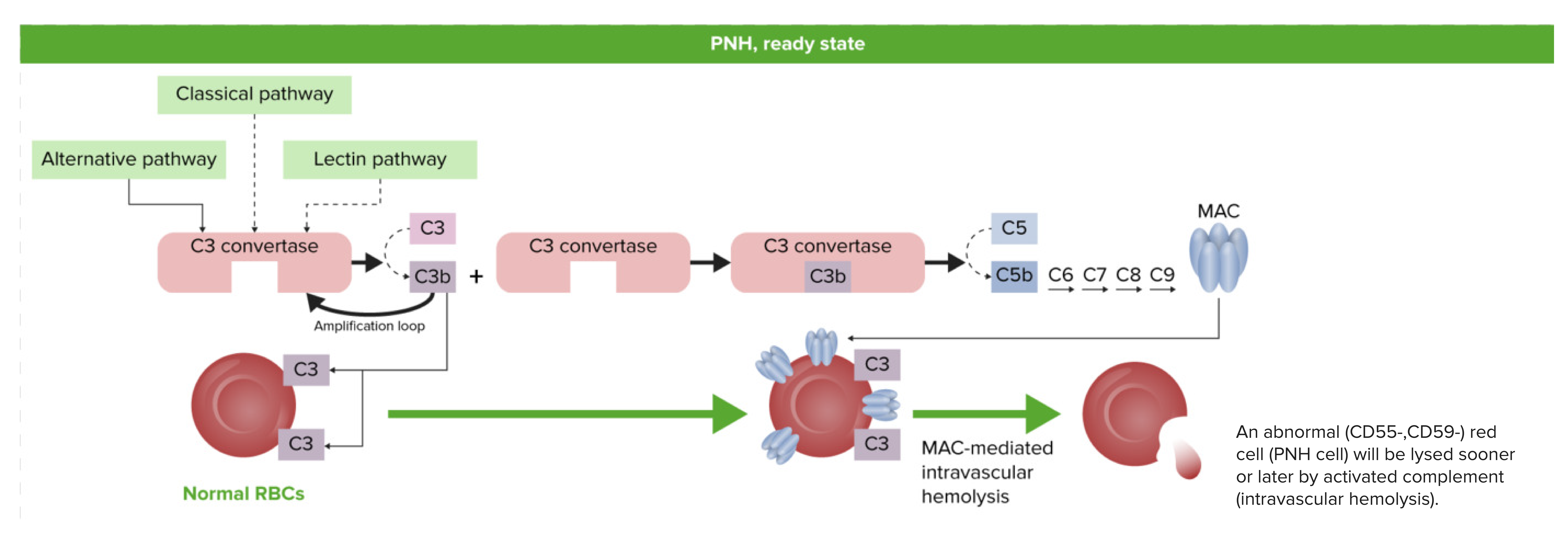
Paroxysmal nocturnal hemoglobinuria (PNH) physiology in the steady state: Even in the absence of inflammation, infection, or other trigger, RBCs with reduced or no GPI on their surfaces are susceptible to complement attack by spontaneous complement activation from a trivial but constantly present amount of activated C3 (the “C3 tickover”). Intravascular hemolysis occurs owing to formation of a membrane attack complex (MAC), which causes focal distortion and holes in the membrane. If extra complement is activated through the classical antigen–antibody pathway, (e.g., by infection), then the hemolysis will be increased; this corresponds to paroxysmal exacerbation of the disease. GPI: glycosylphosphatidylinositol
Bone marrowBone marrowThe soft tissue filling the cavities of bones. Bone marrow exists in two types, yellow and red. Yellow marrow is found in the large cavities of large bones and consists mostly of fat cells and a few primitive blood cells. Red marrow is a hematopoietic tissue and is the site of production of erythrocytes and granular leukocytes. Bone marrow is made up of a framework of connective tissue containing branching fibers with the frame being filled with marrow cells.Bone Marrow: Composition and Hematopoiesis failure
Defined as peripheral cytopeniasCytopeniasIPEX Syndrome associated with deficient hematopoiesisHematopoiesisThe development and formation of various types of blood cells. Hematopoiesis can take place in the bone marrow (medullary) or outside the bone marrow (extramedullary hematopoiesis).Bone Marrow: Composition and Hematopoiesis
Present, to some degree, in all patientsPatientsIndividuals participating in the health care system for the purpose of receiving therapeutic, diagnostic, or preventive procedures.Clinician–Patient Relationship with PNH
Unknown mechanism of association between PNH and aplastic anemiaAplastic AnemiaAplastic anemia (AA) is a rare, life-threatening condition characterized by pancytopenia and hypocellularity of the bone marrow (in the absence of any abnormal cells) reflecting damage to hematopoietic stem cells. Aplastic anemia can be acquired or inherited, however, most cases of AA are acquired and caused by autoimmune damage to hematopoietic stem cells. Aplastic Anemia:
Aplastic anemiaAplastic AnemiaAplastic anemia (AA) is a rare, life-threatening condition characterized by pancytopenia and hypocellularity of the bone marrow (in the absence of any abnormal cells) reflecting damage to hematopoietic stem cells. Aplastic anemia can be acquired or inherited, however, most cases of AA are acquired and caused by autoimmune damage to hematopoietic stem cells. Aplastic Anemia develops in 10%–20% of patientsPatientsIndividuals participating in the health care system for the purpose of receiving therapeutic, diagnostic, or preventive procedures.Clinician–Patient Relationship with PNH.
Paroxysmal nocturnal hemoglobinuriaHemoglobinuriaThe presence of free hemoglobin in the urine, indicating hemolysis of erythrocytes within the vascular system. After saturating the hemoglobin-binding proteins (haptoglobins), free hemoglobin begins to appear in the urine.Transfusion Reactions develops in 5% of patientsPatientsIndividuals participating in the health care system for the purpose of receiving therapeutic, diagnostic, or preventive procedures.Clinician–Patient Relationship with aplastic anemiaAplastic AnemiaAplastic anemia (AA) is a rare, life-threatening condition characterized by pancytopenia and hypocellularity of the bone marrow (in the absence of any abnormal cells) reflecting damage to hematopoietic stem cells. Aplastic anemia can be acquired or inherited, however, most cases of AA are acquired and caused by autoimmune damage to hematopoietic stem cells. Aplastic Anemia.
Bone marrowBone marrowThe soft tissue filling the cavities of bones. Bone marrow exists in two types, yellow and red. Yellow marrow is found in the large cavities of large bones and consists mostly of fat cells and a few primitive blood cells. Red marrow is a hematopoietic tissue and is the site of production of erythrocytes and granular leukocytes. Bone marrow is made up of a framework of connective tissue containing branching fibers with the frame being filled with marrow cells.Bone Marrow: Composition and Hematopoiesis failure in PNH is highly variableVariableVariables represent information about something that can change. The design of the measurement scales, or of the methods for obtaining information, will determine the data gathered and the characteristics of that data. As a result, a variable can be qualitative or quantitative, and may be further classified into subgroups.Types of Variables:
Mild decrease in hematopoietic stem cellsHematopoietic stem cellsProgenitor cells from which all blood cells derived. They are found primarily in the bone marrow and also in small numbers in the peripheral blood.Bone Marrow: Composition and Hematopoiesis (mild end of spectrum)
Mechanism of bone marrowBone marrowThe soft tissue filling the cavities of bones. Bone marrow exists in two types, yellow and red. Yellow marrow is found in the large cavities of large bones and consists mostly of fat cells and a few primitive blood cells. Red marrow is a hematopoietic tissue and is the site of production of erythrocytes and granular leukocytes. Bone marrow is made up of a framework of connective tissue containing branching fibers with the frame being filled with marrow cells.Bone Marrow: Composition and Hematopoiesis failure in PNH:
Proposed mechanism is complement-mediated destruction of hematopoietic precursor cells.
Still poorly understood
AnemiaAnemiaAnemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types
Circulating RBCsRBCsErythrocytes, or red blood cells (RBCs), are the most abundant cells in the blood. While erythrocytes in the fetus are initially produced in the yolk sac then the liver, the bone marrow eventually becomes the main site of production.Erythrocytes: Histology lack the protective effect of CD59
Complement factors assemble/activate MAC on RBC membrane and lyse the cell:
Hemoglobin released into circulationCirculationThe movement of the blood as it is pumped through the cardiovascular system.ABCDE Assessment
Circulating RBCsRBCsErythrocytes, or red blood cells (RBCs), are the most abundant cells in the blood. While erythrocytes in the fetus are initially produced in the yolk sac then the liver, the bone marrow eventually becomes the main site of production.Erythrocytes: Histology lack the protective effect of CD55.
Complement opsonizes or accumulates on red cell membraneCell MembraneA cell membrane (also known as the plasma membrane or plasmalemma) is a biological membrane that separates the cell contents from the outside environment. A cell membrane is composed of a phospholipid bilayer and proteins that function to protect cellular DNA and mediate the exchange of ions and molecules. The Cell: Cell Membrane.
Opsonization marks red cells for phagocytic clearance in the spleenSpleenThe spleen is the largest lymphoid organ in the body, located in the LUQ of the abdomen, superior to the left kidney and posterior to the stomach at the level of the 9th-11th ribs just below the diaphragm. The spleen is highly vascular and acts as an important blood filter, cleansing the blood of pathogens and damaged erythrocytes. Spleen: Anatomy.
Other factors contributing to anemiaAnemiaAnemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types:
IronIronA metallic element with atomic symbol fe, atomic number 26, and atomic weight 55. 85. It is an essential constituent of hemoglobins; cytochromes; and iron-binding proteins. It plays a role in cellular redox reactions and in the transport of oxygen.Trace Elements loss from excessive and chronic red cell lysis:
Some ironIronA metallic element with atomic symbol fe, atomic number 26, and atomic weight 55. 85. It is an essential constituent of hemoglobins; cytochromes; and iron-binding proteins. It plays a role in cellular redox reactions and in the transport of oxygen.Trace Elements lost to hemoglobinuriaHemoglobinuriaThe presence of free hemoglobin in the urine, indicating hemolysis of erythrocytes within the vascular system. After saturating the hemoglobin-binding proteins (haptoglobins), free hemoglobin begins to appear in the urine.Transfusion Reactions
Some ironIronA metallic element with atomic symbol fe, atomic number 26, and atomic weight 55. 85. It is an essential constituent of hemoglobins; cytochromes; and iron-binding proteins. It plays a role in cellular redox reactions and in the transport of oxygen.Trace Elements lost to tissue deposition
Lack of erythrocyte production:
Underlying bone marrowBone marrowThe soft tissue filling the cavities of bones. Bone marrow exists in two types, yellow and red. Yellow marrow is found in the large cavities of large bones and consists mostly of fat cells and a few primitive blood cells. Red marrow is a hematopoietic tissue and is the site of production of erythrocytes and granular leukocytes. Bone marrow is made up of a framework of connective tissue containing branching fibers with the frame being filled with marrow cells.Bone Marrow: Composition and Hematopoiesis failure
IronIronA metallic element with atomic symbol fe, atomic number 26, and atomic weight 55. 85. It is an essential constituent of hemoglobins; cytochromes; and iron-binding proteins. It plays a role in cellular redox reactions and in the transport of oxygen.Trace Elements deficiency
Urine sample from a patient with PNH (left) and from a healthy control (right). The patient’s urine is red because of intravascular hemolysis with hemoglobinuria. Hemolysis occurs throughout the day, not just at night.
Image: “Paroxysmal nocturnal hemoglobinuria in systemic lupus erythematosus: a case report” by Nakamura, N., et al. License: CC BY 2.0
Neutrophil involvement
NeutrophilsNeutrophilsGranular leukocytes having a nucleus with three to five lobes connected by slender threads of chromatin, and cytoplasm containing fine inconspicuous granules and stainable by neutral dyes.Innate Immunity: Phagocytes and Antigen Presentation also exhibit deficiency of GPI-APs:
Increased sensitivity to complement-mediated lysis
Once lysed, may release cytokinesCytokinesNon-antibody proteins secreted by inflammatory leukocytes and some non-leukocytic cells, that act as intercellular mediators. They differ from classical hormones in that they are produced by a number of tissue or cell types rather than by specialized glands. They generally act locally in a paracrine or autocrine rather than endocrine manner.Adaptive Immune Response into circulationCirculationThe movement of the blood as it is pumped through the cardiovascular system.ABCDE Assessment:
Further accelerate complement activity
Increased thrombogenic potential
Excessive neutrophil lysis may lead to increased susceptibility to infection.
Platelet involvement
Platelet-mediated factors include:
Nitrous oxideNitrous oxideNitrogen oxide (N2O). A colorless, odorless gas that is used as an anesthetic and analgesic. High concentrations cause a narcotic effect and may replace oxygen, causing death by asphyxia.Inhaled Anesthetics (NO) deficiency leads to platelet activationPlatelet activationA series of progressive, overlapping events, triggered by exposure of the platelets to subendothelial tissue. These events include shape change, adhesiveness, aggregation, and release reactions. When carried through to completion, these events lead to the formation of a stable hemostatic plug.Hemostasis and aggregationAggregationThe attachment of platelets to one another. This clumping together can be induced by a number of agents (e.g., thrombin; collagen) and is part of the mechanism leading to the formation of a thrombus.Coagulation Studies.
PlateletsPlateletsPlatelets are small cell fragments involved in hemostasis. Thrombopoiesis takes place primarily in the bone marrow through a series of cell differentiation and is influenced by several cytokines. Platelets are formed after fragmentation of the megakaryocyte cytoplasm. Platelets: Histology deficient in GPI-linked proteinsProteinsLinear polypeptides that are synthesized on ribosomes and may be further modified, crosslinked, cleaved, or assembled into complex proteins with several subunits. The specific sequence of amino acids determines the shape the polypeptide will take, during protein folding, and the function of the protein.Energy Homeostasis become activated by complement.
Lack of CD59 on platelet membranes induces platelet aggregationPlatelet aggregationThe attachment of platelets to one another. This clumping together can be induced by a number of agents (e.g., thrombin; collagen) and is part of the mechanism leading to the formation of a thrombus.Hemostasis in particular.
ThrombophiliaThrombophiliaA disorder of hemostasis in which there is a tendency for the occurrence of thrombosis.Hypercoagulable States and thrombosisThrombosisFormation and development of a thrombus or blood clot in the blood vessel.Epidemic Typhus
ThrombosisThrombosisFormation and development of a thrombus or blood clot in the blood vessel.Epidemic Typhus in unusual sites (hepatic, mesenteric, cerebral, dermal veinsVeinsVeins are tubular collections of cells, which transport deoxygenated blood and waste from the capillary beds back to the heart. Veins are classified into 3 types: small veins/venules, medium veins, and large veins. Each type contains 3 primary layers: tunica intima, tunica media, and tunica adventitia. Veins: Histology) is typical of PNH.
NO-mediated factors:
On red cell lysis, hemoglobin released into the circulationCirculationThe movement of the blood as it is pumped through the cardiovascular system.ABCDE Assessment
Hemoglobin is bound to haptoglobin until haptoglobin is saturated.
After saturating haptoglobin, free hemoglobin binds to circulating NO, which:
Normally decreases endothelial adhesionAdhesionThe process whereby platelets adhere to something other than platelets, e.g., collagen; basement membrane; microfibrils; or other ‘foreign’ surfaces.Coagulation Studies
Normally prevents platelet activationPlatelet activationA series of progressive, overlapping events, triggered by exposure of the platelets to subendothelial tissue. These events include shape change, adhesiveness, aggregation, and release reactions. When carried through to completion, these events lead to the formation of a stable hemostatic plug.Hemostasis and aggregationAggregationThe attachment of platelets to one another. This clumping together can be induced by a number of agents (e.g., thrombin; collagen) and is part of the mechanism leading to the formation of a thrombus.Coagulation Studies
Normally causes vasodilationVasodilationThe physiological widening of blood vessels by relaxing the underlying vascular smooth muscle.Pulmonary Hypertension Drugs
But if NO is depleted in the peripheral circulationCirculationThe movement of the blood as it is pumped through the cardiovascular system.ABCDE Assessment:
↓ NO enhances endothelial adhesionAdhesionThe process whereby platelets adhere to something other than platelets, e.g., collagen; basement membrane; microfibrils; or other ‘foreign’ surfaces.Coagulation Studies.
↓ NO enhances platelet activationPlatelet activationA series of progressive, overlapping events, triggered by exposure of the platelets to subendothelial tissue. These events include shape change, adhesiveness, aggregation, and release reactions. When carried through to completion, these events lead to the formation of a stable hemostatic plug.Hemostasis and aggregationAggregationThe attachment of platelets to one another. This clumping together can be induced by a number of agents (e.g., thrombin; collagen) and is part of the mechanism leading to the formation of a thrombus.Coagulation Studies.
↓ NO causes vasoconstrictionVasoconstrictionThe physiological narrowing of blood vessels by contraction of the vascular smooth muscle.Vascular Resistance, Flow, and Mean Arterial Pressure → exposure of tissue factor → triggers the extrinsic coagulation cascadeCoagulation cascadeThe coagulation cascade is a series of reactions that ultimately generates a strong, cross-linked fibrin clot.Hemostasis.
Platelet-mediated factors:
NO deficiency leads to platelet activationPlatelet activationA series of progressive, overlapping events, triggered by exposure of the platelets to subendothelial tissue. These events include shape change, adhesiveness, aggregation, and release reactions. When carried through to completion, these events lead to the formation of a stable hemostatic plug.Hemostasis and aggregationAggregationThe attachment of platelets to one another. This clumping together can be induced by a number of agents (e.g., thrombin; collagen) and is part of the mechanism leading to the formation of a thrombus.Coagulation Studies.
PlateletsPlateletsPlatelets are small cell fragments involved in hemostasis. Thrombopoiesis takes place primarily in the bone marrow through a series of cell differentiation and is influenced by several cytokines. Platelets are formed after fragmentation of the megakaryocyte cytoplasm. Platelets: Histology deficient in GPI-linked proteinsProteinsLinear polypeptides that are synthesized on ribosomes and may be further modified, crosslinked, cleaved, or assembled into complex proteins with several subunits. The specific sequence of amino acids determines the shape the polypeptide will take, during protein folding, and the function of the protein.Energy Homeostasis become activated by complement.
Lack of CD59 on platelet membranes induces platelet aggregationPlatelet aggregationThe attachment of platelets to one another. This clumping together can be induced by a number of agents (e.g., thrombin; collagen) and is part of the mechanism leading to the formation of a thrombus.Hemostasis.
Vascular factors:
Activation of complement on endothelial cell surfaces
Slow flowFlowBlood flows through the heart, arteries, capillaries, and veins in a closed, continuous circuit. Flow is the movement of volume per unit of time. Flow is affected by the pressure gradient and the resistance fluid encounters between 2 points. Vascular resistance is the opposition to flow, which is caused primarily by blood friction against vessel walls.Vascular Resistance, Flow, and Mean Arterial Pressure: PNH thrombosisThrombosisFormation and development of a thrombus or blood clot in the blood vessel.Epidemic Typhus is far more common in the venous than in the arterial system, notably in large veinsLarge veinsVeins: Histology or where flowFlowBlood flows through the heart, arteries, capillaries, and veins in a closed, continuous circuit. Flow is the movement of volume per unit of time. Flow is affected by the pressure gradient and the resistance fluid encounters between 2 points. Vascular resistance is the opposition to flow, which is caused primarily by blood friction against vessel walls.Vascular Resistance, Flow, and Mean Arterial Pressure is slowest.
Hepatic
Portal
Splenic
Mesenteric
Pulmonary
Cerebral
Dermal
Other factors:
Unknown mechanism of increased procoagulant and fibrinolytic activity
White cell lysis may release vasoactive and/or thrombogenic molecules.
Renal involvement
Renal failureRenal failureConditions in which the kidneys perform below the normal level in the ability to remove wastes, concentrate urine, and maintain electrolyte balance; blood pressure; and calcium metabolism. Renal insufficiency can be classified by the degree of kidney damage (as measured by the level of proteinuria) and reduction in glomerular filtration rate.Crush Syndrome is progressive and multifactorial.
Acute tubular necrosisNecrosisThe death of cells in an organ or tissue due to disease, injury or failure of the blood supply.Ischemic Cell Damage effects of heme and ironIronA metallic element with atomic symbol fe, atomic number 26, and atomic weight 55. 85. It is an essential constituent of hemoglobins; cytochromes; and iron-binding proteins. It plays a role in cellular redox reactions and in the transport of oxygen.Trace Elements (pigment nephropathy)
Tubular obstruction with pigment casts
Decreased renal perfusion from renal veinRenal veinShort thick veins which return blood from the kidneys to the vena cava.Glomerular FiltrationthrombosisThrombosisFormation and development of a thrombus or blood clot in the blood vessel.Epidemic Typhus
Smooth muscle dystoniaDystoniaDystonia is a hyperkinetic movement disorder characterized by the involuntary contraction of muscles, resulting in abnormal postures or twisting and repetitive movements. Dystonia can present in various ways as may affect many different skeletal muscle groups. Dystonia
Upon red cell lysis, hemoglobin released into the circulationCirculationThe movement of the blood as it is pumped through the cardiovascular system.ABCDE Assessment
Hemoglobin is bound to haptoglobin until haptoglobin is saturated.
After saturating haptoglobin, free hemoglobin binds to circulating NO, which:
Normally relaxes smooth muscle toneMuscle toneThe state of activity or tension of a muscle beyond that related to its physical properties, that is, its active resistance to stretch. In skeletal muscle, tonus is dependent upon efferent innervation.Skeletal Muscle Contraction
Is depleted in the peripheral circulationCirculationThe movement of the blood as it is pumped through the cardiovascular system.ABCDE Assessment
NO depletion leads to smooth muscle contractionSmooth muscle contractionSmooth muscle is primarily found in the walls of hollow structures and some visceral organs, including the walls of the vasculature, GI, respiratory, and genitourinary tracts. Smooth muscle contracts more slowly and is regulated differently than skeletal muscle. Smooth muscle can be stimulated by nerve impulses, hormones, metabolic factors (like pH, CO2 or O2 levels), its own intrinsic pacemaker ability, or even mechanical stretch.Smooth Muscle Contraction.
Clinical manifestations depend on the smooth muscle population affected.
Paroxysms of hemolysis
Exacerbations of hemolysis may be due to:
Administration of ironIronA metallic element with atomic symbol fe, atomic number 26, and atomic weight 55. 85. It is an essential constituent of hemoglobins; cytochromes; and iron-binding proteins. It plays a role in cellular redox reactions and in the transport of oxygen.Trace Elements to an iron-deficient patient:
ErythrocytesErythrocytesErythrocytes, or red blood cells (RBCs), are the most abundant cells in the blood. While erythrocytes in the fetus are initially produced in the yolk sac then the liver, the bone marrow eventually becomes the main site of production. Erythrocytes: Histology lacking CD59/CD55 released into circulationCirculationThe movement of the blood as it is pumped through the cardiovascular system.ABCDE Assessment
Exogenous activation of the complement systemComplement systemSerum glycoproteins participating in the host defense mechanism of complement activation that creates the complement membrane attack complex. Included are glycoproteins in the various pathways of complement activation (classical complement pathway; alternative complement pathway; and lectin complement pathway).Innate Immunity: Barriers, Complement, and Cytokines:
Infection/sepsisSepsisSystemic inflammatory response syndrome with a proven or suspected infectious etiology. When sepsis is associated with organ dysfunction distant from the site of infection, it is called severe sepsis. When sepsis is accompanied by hypotension despite adequate fluid infusion, it is called septic shock.Sepsis and Septic Shock
Surgery/trauma
Drugs
Antibody involvement
Coombs testing detects the presence of antibodiesAntibodiesImmunoglobulins (Igs), also known as antibodies, are glycoprotein molecules produced by plasma cells that act in immune responses by recognizing and binding particular antigens. The various Ig classes are IgG (the most abundant), IgM, IgE, IgD, and IgA, which differ in their biologic features, structure, target specificity, and distribution.Immunoglobulins: Types and Functions in autoimmune hemolytic anemias.
Untreated PNH is a Coombs-negative hemolytic anemiaHemolytic AnemiaHemolytic anemia (HA) is the term given to a large group of anemias that are caused by the premature destruction/hemolysis of circulating red blood cells (RBCs). Hemolysis can occur within (intravascular hemolysis) or outside the blood vessels (extravascular hemolysis). Hemolytic Anemia.
Coombs-positivity often occurs in patientsPatientsIndividuals participating in the health care system for the purpose of receiving therapeutic, diagnostic, or preventive procedures.Clinician–Patient Relationship with PNH treated with eculizumab:
Eculizumab is a monoclonal antibody that blocks complement-mediated formation of MAC:
Complement factors not consumed by MAC formation are free in the circulationCirculationThe movement of the blood as it is pumped through the cardiovascular system.ABCDE Assessment and coat red cell surfaces lacking protection from CD55:
RBC is now opsonized by complement.
Opsonized RBCsRBCsErythrocytes, or red blood cells (RBCs), are the most abundant cells in the blood. While erythrocytes in the fetus are initially produced in the yolk sac then the liver, the bone marrow eventually becomes the main site of production.Erythrocytes: Histology undergo extravascular hemolysisExtravascular hemolysisHemolytic Anemia.
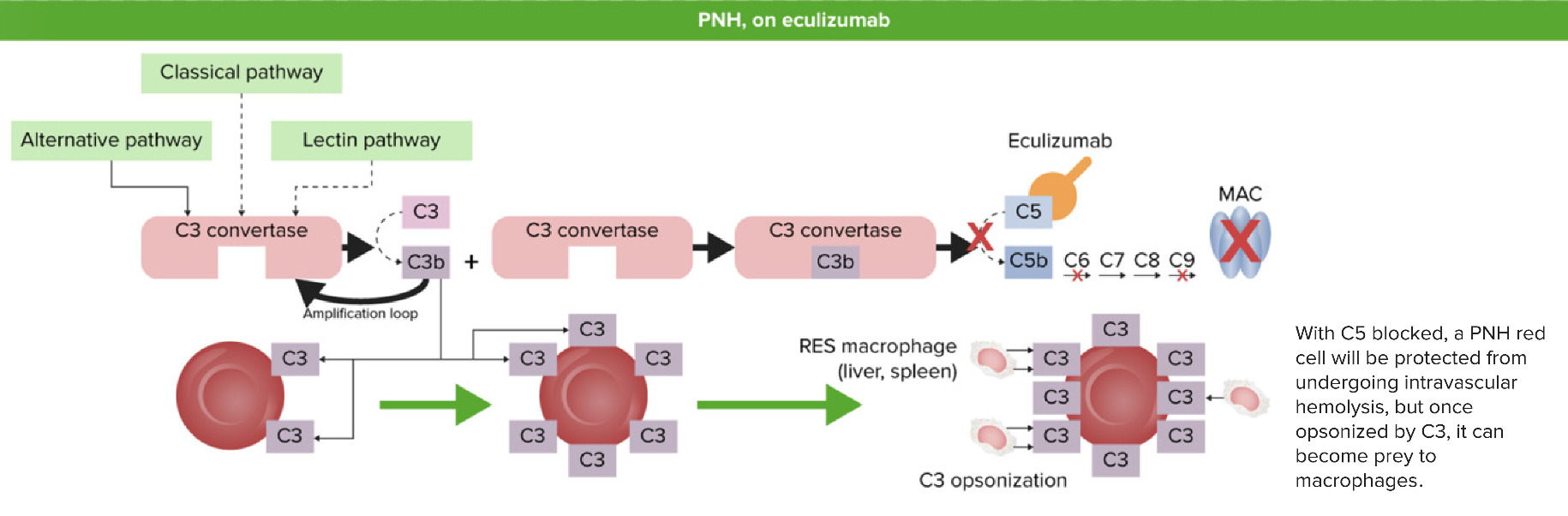
Paroxysmal nocturnal hemoglobinuria after eculizumab: When C5 is blocked (inhibited) by eculizumab, C5 convertase cannot form C5b from the cleavage of C5. Membrane attack complex (MAC) is not formed and no intravascular hemolysis occurs. However, the C3 fragments that were released earlier in the activation process will coat the unprotected RBCs and cause extravascular hemolysis by the reticuloendothelial system (RES) in the liver and spleen.
HemoglobinuriaHemoglobinuriaThe presence of free hemoglobin in the urine, indicating hemolysis of erythrocytes within the vascular system. After saturating the hemoglobin-binding proteins (haptoglobins), free hemoglobin begins to appear in the urine.Transfusion Reactions:
Classically noticed at 1st morning void
Can occur at any time of day or night
Intermittent (paroxysmal) in nature
JaundiceJaundiceJaundice is the abnormal yellowing of the skin and/or sclera caused by the accumulation of bilirubin. Hyperbilirubinemia is caused by either an increase in bilirubin production or a decrease in the hepatic uptake, conjugation, or excretion of bilirubin. Jaundice/icterusIcterusA clinical manifestation of hyperbilirubinemia, characterized by the yellowish staining of the skin, mucous membranes, and sclera. Clinical jaundice usually is a sign of liver dysfunction.Jaundice
Acute renal failureRenal failureConditions in which the kidneys perform below the normal level in the ability to remove wastes, concentrate urine, and maintain electrolyte balance; blood pressure; and calcium metabolism. Renal insufficiency can be classified by the degree of kidney damage (as measured by the level of proteinuria) and reduction in glomerular filtration rate.Crush Syndrome
Chronic renal failureRenal failureConditions in which the kidneys perform below the normal level in the ability to remove wastes, concentrate urine, and maintain electrolyte balance; blood pressure; and calcium metabolism. Renal insufficiency can be classified by the degree of kidney damage (as measured by the level of proteinuria) and reduction in glomerular filtration rate.Crush Syndrome
Anemia-associated symptoms
FatigueFatigueThe state of weariness following a period of exertion, mental or physical, characterized by a decreased capacity for work and reduced efficiency to respond to stimuli.Fibromyalgia
Weakness
DyspneaDyspneaDyspnea is the subjective sensation of breathing discomfort. Dyspnea is a normal manifestation of heavy physical or psychological exertion, but also may be caused by underlying conditions (both pulmonary and extrapulmonary). Dyspnea
TachycardiaTachycardiaAbnormally rapid heartbeat, usually with a heart rate above 100 beats per minute for adults. Tachycardia accompanied by disturbance in the cardiac depolarization (cardiac arrhythmia) is called tachyarrhythmia.Sepsis in Children
Smooth muscle contractionSmooth muscle contractionSmooth muscle is primarily found in the walls of hollow structures and some visceral organs, including the walls of the vasculature, GI, respiratory, and genitourinary tracts. Smooth muscle contracts more slowly and is regulated differently than skeletal muscle. Smooth muscle can be stimulated by nerve impulses, hormones, metabolic factors (like pH, CO2 or O2 levels), its own intrinsic pacemaker ability, or even mechanical stretch.Smooth Muscle Contraction–associated symptoms
These symptoms are due to decreased circulating NO:
DysphagiaDysphagiaDysphagia is the subjective sensation of difficulty swallowing. Symptoms can range from a complete inability to swallow, to the sensation of solids or liquids becoming “stuck.” Dysphagia is classified as either oropharyngeal or esophageal, with esophageal dysphagia having 2 sub-types: functional and mechanical. Dysphagia (esophageal)
NauseaNauseaAn unpleasant sensation in the stomach usually accompanied by the urge to vomit. Common causes are early pregnancy, sea and motion sickness, emotional stress, intense pain, food poisoning, and various enteroviruses.Antiemetics (intestinal)
Erectile dysfunctionErectile DysfunctionErectile dysfunction (ED) is defined as the inability to achieve or maintain a penile erection, resulting in difficulty to perform penetrative sexual intercourse. Local penile factors and systemic diseases, including diabetes, cardiac disease, and neurological disorders, can cause ED. Erectile Dysfunction (microvascular/corpus cavernosa)
Pulmonary hypertensionPulmonary HypertensionPulmonary hypertension (PH) or pulmonary arterial hypertension (PAH) is characterized by elevated pulmonary arterial pressure, which can lead to chronic progressive right heart failure. Pulmonary hypertension is grouped into 5 categories based on etiology, which include primary PAH, and PH due to cardiac disease, lung or hypoxic disease, chronic thromboembolic disease, and multifactorial or unclear etiologies. Pulmonary Hypertension (pulmonary arterial)
Thrombosis-associated symptoms
Limb edemaEdemaEdema is a condition in which excess serous fluid accumulates in the body cavity or interstitial space of connective tissues. Edema is a symptom observed in several medical conditions. It can be categorized into 2 types, namely, peripheral (in the extremities) and internal (in an organ or body cavity). Edema/discoloration/painPainAn unpleasant sensation induced by noxious stimuli which are detected by nerve endings of nociceptive neurons.Pain: Types and Pathways from deep vein thrombosisThrombosisFormation and development of a thrombus or blood clot in the blood vessel.Epidemic Typhus (DVTDVTDeep vein thrombosis (DVT) usually occurs in the deep veins of the lower extremities. The affected veins include the femoral, popliteal, iliofemoral, and pelvic veins. Proximal DVT is more likely to cause a pulmonary embolism (PE) and is generally considered more serious. Deep Vein Thrombosis)
Pulmonary embolismPulmonary EmbolismPulmonary embolism (PE) is a potentially fatal condition that occurs as a result of intraluminal obstruction of the main pulmonary artery or its branches. The causative factors include thrombi, air, amniotic fluid, and fat. In PE, gas exchange is impaired due to the decreased return of deoxygenated blood to the lungs. Pulmonary Embolism
Cerebral vein/dural vein thrombosisThrombosisFormation and development of a thrombus or blood clot in the blood vessel.Epidemic Typhus leads to:
Intracranial hypertensionHypertensionHypertension, or high blood pressure, is a common disease that manifests as elevated systemic arterial pressures. Hypertension is most often asymptomatic and is found incidentally as part of a routine physical examination or during triage for an unrelated medical encounter. Hypertension
Dermal vein thrombosisThrombosisFormation and development of a thrombus or blood clot in the blood vessel.Epidemic Typhus leads to:
Bruising
Purpura fulminansPurpura fulminansA severe, rapidly fatal reaction occurring most commonly in children following an infectious illness. It is characterized by large, rapidly spreading skin hemorrhages, fever, or shock. Purpura fulminans often accompanies or is triggered by disseminated intravascular coagulation.Disseminated Intravascular Coagulation
Hepatic/portal/mesenteric vein thrombosisThrombosisFormation and development of a thrombus or blood clot in the blood vessel.Epidemic Typhus leads to:
SplenomegalySplenomegalySplenomegaly is pathologic enlargement of the spleen that is attributable to numerous causes, including infections, hemoglobinopathies, infiltrative processes, and outflow obstruction of the portal vein. Splenomegaly
Budd-Chiari syndromeBudd-Chiari syndromeBudd-Chiari syndrome is a condition resulting from the interruption of the normal outflow of blood from the liver. The primary type arises from a venous process (affecting the hepatic veins or inferior vena cava) such as thrombosis, but can also be from a lesion compressing or invading the veins (secondary type). The patient typically presents with hepatomegaly, ascites, and abdominal discomfort. Budd-Chiari Syndrome
Neutropenia-associated symptoms
Susceptibility to infection
Increased circulating cytokinesCytokinesNon-antibody proteins secreted by inflammatory leukocytes and some non-leukocytic cells, that act as intercellular mediators. They differ from classical hormones in that they are produced by a number of tissue or cell types rather than by specialized glands. They generally act locally in a paracrine or autocrine rather than endocrine manner.Adaptive Immune Response leads to:
MalaiseMalaiseTick-borne Encephalitis Virus/fatigueFatigueThe state of weariness following a period of exertion, mental or physical, characterized by a decreased capacity for work and reduced efficiency to respond to stimuli.Fibromyalgia
InflammationInflammationInflammation is a complex set of responses to infection and injury involving leukocytes as the principal cellular mediators in the body’s defense against pathogenic organisms. Inflammation is also seen as a response to tissue injury in the process of wound healing. The 5 cardinal signs of inflammation are pain, heat, redness, swelling, and loss of function. Inflammation
Sepsis-like syndrome
Diagnosis
CBC
AnemiaAnemiaAnemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types (↓ RBC count, ↓ hematocritHematocritThe volume of packed red blood cells in a blood specimen. The volume is measured by centrifugation in a tube with graduated markings, or with automated blood cell counters. It is an indicator of erythrocyte status in disease. For example, anemia shows a low value; polycythemia, a high value.Neonatal Polycythemia):
VariableVariableVariables represent information about something that can change. The design of the measurement scales, or of the methods for obtaining information, will determine the data gathered and the characteristics of that data. As a result, a variable can be qualitative or quantitative, and may be further classified into subgroups.Types of Variables, but generally present to some degree
May be subtle or pronounced depending on degree of hemolysis
May or may not be normocytic, microcytic, or macrocytic depending on degree of vitamin B12, folateFolateFolate and vitamin B12 are 2 of the most clinically important water-soluble vitamins. Deficiencies can present with megaloblastic anemia, GI symptoms, neuropsychiatric symptoms, and adverse pregnancy complications, including neural tube defects. Folate and Vitamin B12, or ironIronA metallic element with atomic symbol fe, atomic number 26, and atomic weight 55. 85. It is an essential constituent of hemoglobins; cytochromes; and iron-binding proteins. It plays a role in cellular redox reactions and in the transport of oxygen.Trace Elements deficiency
↓ or normal neutrophil count
↓ or normal platelet count
↑ Free hemoglobin
Chemistry
↑ ASTASTEnzymes of the transferase class that catalyze the conversion of l-aspartate and 2-ketoglutarate to oxaloacetate and l-glutamate.Liver Function Tests/ALTALTAn enzyme that catalyzes the conversion of l-alanine and 2-oxoglutarate to pyruvate and l-glutamate.Liver Function Tests (if hepatic congestion)
↓ Leukocyte alkaline phosphataseAlkaline PhosphataseAn enzyme that catalyzes the conversion of an orthophosphoric monoester and water to an alcohol and orthophosphate.Osteosarcoma (LAP)
UrinalysisUrinalysisExamination of urine by chemical, physical, or microscopic means. Routine urinalysis usually includes performing chemical screening tests, determining specific gravity, observing any unusual color or odor, screening for bacteriuria, and examining the sediment microscopically.Urinary Tract Infections (UTIs) in Children
Gross red or pink discoloration
Positive for hemoglobin
No RBCsRBCsErythrocytes, or red blood cells (RBCs), are the most abundant cells in the blood. While erythrocytes in the fetus are initially produced in the yolk sac then the liver, the bone marrow eventually becomes the main site of production.Erythrocytes: Histology on microscopy
+/– protein depending on renal integrity
Bone marrowBone marrowThe soft tissue filling the cavities of bones. Bone marrow exists in two types, yellow and red. Yellow marrow is found in the large cavities of large bones and consists mostly of fat cells and a few primitive blood cells. Red marrow is a hematopoietic tissue and is the site of production of erythrocytes and granular leukocytes. Bone marrow is made up of a framework of connective tissue containing branching fibers with the frame being filled with marrow cells.Bone Marrow: Composition and Hematopoiesis analysis
GPI-AP deficiency can be demonstrated on bone marrowBone marrowThe soft tissue filling the cavities of bones. Bone marrow exists in two types, yellow and red. Yellow marrow is found in the large cavities of large bones and consists mostly of fat cells and a few primitive blood cells. Red marrow is a hematopoietic tissue and is the site of production of erythrocytes and granular leukocytes. Bone marrow is made up of a framework of connective tissue containing branching fibers with the frame being filled with marrow cells.Bone Marrow: Composition and Hematopoiesis cells using flow cytometryFlow cytometryTechnique using an instrument system for making, processing, and displaying one or more measurements on individual cells obtained from a cell suspension. Cells are usually stained with one or more fluorescent dyes specific to cell components of interest, e.g., DNA, and fluorescence of each cell is measured as it rapidly transverses the excitation beam (laser or mercury arc lamp). Fluorescence provides a quantitative measure of various biochemical and biophysical properties of the cell, as well as a basis for cell sorting. Other measurable optical parameters include light absorption and light scattering, the latter being applicable to the measurement of cell size, shape, density, granularity, and stain uptake.X-linked Agammaglobulinemia.
Unnecessary for standard diagnosis of PNH
Needed for proper classification:
Evaluate for other marrow failure syndromes.
Evaluate for other clonal myelopathies.
Flow cytometryFlow cytometryTechnique using an instrument system for making, processing, and displaying one or more measurements on individual cells obtained from a cell suspension. Cells are usually stained with one or more fluorescent dyes specific to cell components of interest, e.g., DNA, and fluorescence of each cell is measured as it rapidly transverses the excitation beam (laser or mercury arc lamp). Fluorescence provides a quantitative measure of various biochemical and biophysical properties of the cell, as well as a basis for cell sorting. Other measurable optical parameters include light absorption and light scattering, the latter being applicable to the measurement of cell size, shape, density, granularity, and stain uptake.X-linked Agammaglobulinemia (FCM) performed on peripheral blood
HemoglobinuriaHemoglobinuriaThe presence of free hemoglobin in the urine, indicating hemolysis of erythrocytes within the vascular system. After saturating the hemoglobin-binding proteins (haptoglobins), free hemoglobin begins to appear in the urine.Transfusion Reactions
Abnormally high serum LDHLDHOsteosarcoma, especially with concurrent ironIronA metallic element with atomic symbol fe, atomic number 26, and atomic weight 55. 85. It is an essential constituent of hemoglobins; cytochromes; and iron-binding proteins. It plays a role in cellular redox reactions and in the transport of oxygen.Trace Elements deficiency
Aplastic anemiaAplastic AnemiaAplastic anemia (AA) is a rare, life-threatening condition characterized by pancytopenia and hypocellularity of the bone marrow (in the absence of any abnormal cells) reflecting damage to hematopoietic stem cells. Aplastic anemia can be acquired or inherited, however, most cases of AA are acquired and caused by autoimmune damage to hematopoietic stem cells. Aplastic Anemia (at diagnosis and once yearly)
Refractory anemiaAnemiaAnemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types
Myelodysplastic syndrome
Episodic dysphagiaDysphagiaDysphagia is the subjective sensation of difficulty swallowing. Symptoms can range from a complete inability to swallow, to the sensation of solids or liquids becoming “stuck.” Dysphagia is classified as either oropharyngeal or esophageal, with esophageal dysphagia having 2 sub-types: functional and mechanical. Dysphagia or abdominal painAbdominal PainAcute Abdomen with evidence of intravascular hemolysisIntravascular hemolysisHemolytic Anemia
Indicated for definitive diagnosis and monitoring of PNH:
Must show ≥ 5% population of RBCsRBCsErythrocytes, or red blood cells (RBCs), are the most abundant cells in the blood. While erythrocytes in the fetus are initially produced in the yolk sac then the liver, the bone marrow eventually becomes the main site of production.Erythrocytes: Histology that are deficient in CD55 and CD59 or ≥ 20% of CD55/CD59-deficient granulocytesGranulocytesLeukocytes with abundant granules in the cytoplasm. They are divided into three groups according to the staining properties of the granules: neutrophilic, eosinophilic, and basophilic. Mature granulocytes are the neutrophils; eosinophils; and basophils.White Myeloid Cells: Histology to be diagnostic for PNH.
PatientsPatientsIndividuals participating in the health care system for the purpose of receiving therapeutic, diagnostic, or preventive procedures.Clinician–Patient Relationship with proven GPI-AP deficiency should be monitored yearly to track size of clonal population.
EliminationEliminationThe initial damage and destruction of tumor cells by innate and adaptive immunity. Completion of the phase means no cancer growth. Cancer Immunotherapy or marked reduction in blood transfusionsBlood transfusionsThe introduction of whole blood or blood component directly into the bloodstream.Transfusion Products
Marked decrease in thrombotic complications
Administration:
IV every 2 weeks for eculizumab
IV every 8 weeks for ravulizumab
Disadvantages:
Increased risk of life-threatening NeisseriaNeisseriaNeisseria is a genus of bacteria commonly present on mucosal surfaces. Several species exist, but only 2 are pathogenic to humans: N. gonorrhoeae and N. meningitidis. Neisseria species are non-motile, gram-negative diplococci most commonly isolated on modified Thayer-Martin (MTM) agar. NeisseriainfectionsInfectionsInvasion of the host organism by microorganisms or their toxins or by parasites that can cause pathological conditions or diseases.Chronic Granulomatous Disease, requiring pretreatment vaccinationVaccinationVaccination is the administration of a substance to induce the immune system to develop protection against a disease. Unlike passive immunization, which involves the administration of pre-performed antibodies, active immunization constitutes the administration of a vaccine to stimulate the body to produce its own antibodies.Vaccination and daily antibiotics for prophylaxisProphylaxisCephalosporins
High cost: approximately $400,000 (U.S.)/year
Extravascular hemolysisExtravascular hemolysisHemolytic Anemia will still continue, owing to opsonization of RBCsRBCsErythrocytes, or red blood cells (RBCs), are the most abundant cells in the blood. While erythrocytes in the fetus are initially produced in the yolk sac then the liver, the bone marrow eventually becomes the main site of production.Erythrocytes: Histology by C3 fragments.
Bone marrow transplantationBone marrow transplantationTransfer of hematopoietic stem cells from bone marrow or blood between individuals within the same species (homologous transplantation) or transfer within the same individual (autologous transplantation). Hematopoietic stem cell transplantation has been used as an alternative to bone marrow transplantation in the treatment of a variety of neoplasms.Organ Transplantation
The only definitive cure, but has substantial intrinsic risks
PNH complications unresponsive to complement inhibitors (or if they are not available)
Supportive care
Always provided regardless of other therapies provided
RBC transfusions of leukocyte-reduced/filtered blood (used now routinely for all transfused blood):
Decreases risk of antibody reaction to leukocyte antigens
Decreases complement activationComplement ActivationThe sequential activation of serum complement proteins to create the complement membrane attack complex. Factors initiating complement activation include antigen-antibody complexes, microbial antigens, or cell surface polysaccharides.Systemic Lupus Erythematosus
Vitamin B12/folic acid supplementation if ongoing hemolysis
Iron supplementationIron SupplementationIron Deficiency Anemia if ironIronA metallic element with atomic symbol fe, atomic number 26, and atomic weight 55. 85. It is an essential constituent of hemoglobins; cytochromes; and iron-binding proteins. It plays a role in cellular redox reactions and in the transport of oxygen.Trace Elements deficient owing to hemoglobinuriaHemoglobinuriaThe presence of free hemoglobin in the urine, indicating hemolysis of erythrocytes within the vascular system. After saturating the hemoglobin-binding proteins (haptoglobins), free hemoglobin begins to appear in the urine.Transfusion Reactions
AnticoagulantsAnticoagulantsAnticoagulants are drugs that retard or interrupt the coagulation cascade. The primary classes of available anticoagulants include heparins, vitamin K-dependent antagonists (e.g., warfarin), direct thrombin inhibitors, and factor Xa inhibitors. Anticoagulants in patientsPatientsIndividuals participating in the health care system for the purpose of receiving therapeutic, diagnostic, or preventive procedures.Clinician–Patient Relationship with history of or genetic predisposition to thrombosisThrombosisFormation and development of a thrombus or blood clot in the blood vessel.Epidemic Typhus: lifelong anticoagulationAnticoagulationPulmonary Hypertension Drugs if high risk or if atypical thrombosisThrombosisFormation and development of a thrombus or blood clot in the blood vessel.Epidemic Typhus has occurred
ReticulocyteReticulocyteImmature erythrocytes. In humans, these are erythroid cells that have just undergone extrusion of their cell nucleus. They still contain some organelles that gradually decrease in number as the cells mature. Ribosomes are last to disappear. Certain staining techniques cause components of the ribosomes to precipitate into characteristic ‘reticulum’ (not the same as the endoplasmic reticulum), hence the name reticulocytes.Erythrocytes: Histology count
Chemistry profile (electrolytesElectrolytesElectrolytes are mineral salts that dissolve in water and dissociate into charged particles called ions, which can be either be positively (cations) or negatively (anions) charged. Electrolytes are distributed in the extracellular and intracellular compartments in different concentrations. Electrolytes are essential for various basic life-sustaining functions.Electrolytes, renal function, hepatic function)
Iron studiesIron StudiesIron Deficiency Anemia (serum ironIronA metallic element with atomic symbol fe, atomic number 26, and atomic weight 55. 85. It is an essential constituent of hemoglobins; cytochromes; and iron-binding proteins. It plays a role in cellular redox reactions and in the transport of oxygen.Trace Elements, total ironIronA metallic element with atomic symbol fe, atomic number 26, and atomic weight 55. 85. It is an essential constituent of hemoglobins; cytochromes; and iron-binding proteins. It plays a role in cellular redox reactions and in the transport of oxygen.Trace Elements binding capacity, and ferritinFerritinIron-containing proteins that are widely distributed in animals, plants, and microorganisms. Their major function is to store iron in a nontoxic bioavailable form. Each ferritin molecule consists of ferric iron in a hollow protein shell (apoferritins) made of 24 subunits of various sequences depending on the species and tissue types.Hereditary Hemochromatosis)
FCM to determine any change in PNH clone size
Bone marrowBone marrowThe soft tissue filling the cavities of bones. Bone marrow exists in two types, yellow and red. Yellow marrow is found in the large cavities of large bones and consists mostly of fat cells and a few primitive blood cells. Red marrow is a hematopoietic tissue and is the site of production of erythrocytes and granular leukocytes. Bone marrow is made up of a framework of connective tissue containing branching fibers with the frame being filled with marrow cells.Bone Marrow: Composition and HematopoiesisbiopsyBiopsyRemoval and pathologic examination of specimens from the living body.Ewing Sarcoma if pancytopeniaPancytopeniaDeficiency of all three cell elements of the blood, erythrocytes, leukocytes and platelets.Aplastic Anemia or suspected aplastic anemiaAplastic AnemiaAplastic anemia (AA) is a rare, life-threatening condition characterized by pancytopenia and hypocellularity of the bone marrow (in the absence of any abnormal cells) reflecting damage to hematopoietic stem cells. Aplastic anemia can be acquired or inherited, however, most cases of AA are acquired and caused by autoimmune damage to hematopoietic stem cells. Aplastic Anemia or myelodysplastic syndrome
Prognosis
Natural history, with only supportive treatment given:
Median survival after diagnosis: 10–20 years
Major complications:
PancytopeniaPancytopeniaDeficiency of all three cell elements of the blood, erythrocytes, leukocytes and platelets.Aplastic Anemia
ThrombosisThrombosisFormation and development of a thrombus or blood clot in the blood vessel.Epidemic Typhus
AMLAMLAcute myeloid leukemia (AML) is a hematologic malignancy characterized by the uncontrolled proliferation of myeloid precursor cells. Seen predominantly in older adults, AML includes an accumulation of myeloblasts and a replacement of normal marrow by malignant cells, which leads to impaired hematopoiesis. Acute Myeloid Leukemia
With complement inhibitors (eculizumab and ravulizumab), mortalityMortalityAll deaths reported in a given population.Measures of Health Status appears similar to that for age-matched controls, but limited time data so far
Bone marrow transplantationBone marrow transplantationTransfer of hematopoietic stem cells from bone marrow or blood between individuals within the same species (homologous transplantation) or transfer within the same individual (autologous transplantation). Hematopoietic stem cell transplantation has been used as an alternative to bone marrow transplantation in the treatment of a variety of neoplasms.Organ Transplantation: curative but significant inherent risks involved
Full, spontaneous recovery rarely occurs.
Differential Diagnosis
Hemolytic anemiaHemolytic AnemiaHemolytic anemia (HA) is the term given to a large group of anemias that are caused by the premature destruction/hemolysis of circulating red blood cells (RBCs). Hemolysis can occur within (intravascular hemolysis) or outside the blood vessels (extravascular hemolysis). Hemolytic Anemia (HAHAHemolytic anemia (HA) is the term given to a large group of anemias that are caused by the premature destruction/hemolysis of circulating red blood cells (RBCs). Hemolysis can occur within (intravascular hemolysis) or outside the blood vessels (extravascular hemolysis).Hemolytic Anemia): hereditary disorders (e.g., glucose-6-phosphate dehydrogenaseGlucose-6-phosphate dehydrogenasePentose Phosphate Pathway (G6PDG6PDPentose Phosphate Pathway) deficiency, hereditary spherocytosisHereditary SpherocytosisHereditary spherocytosis (HS) is the most common type of hereditary hemolytic anemia. The condition is caused by a cytoskeletal protein deficiency in the RBC membrane. This results in loss of membrane stability and deformability of the RBC, giving the cell its spherical shape (spherocyte). Hereditary Spherocytosis, sickle cell anemiaSickle cell anemiaA disease characterized by chronic hemolytic anemia, episodic painful crises, and pathologic involvement of many organs. It is the clinical expression of homozygosity for hemoglobin S.Sickle Cell Disease) or acquired disorders (e.g., immune disorders, toxic chemicals and drugs, antiviralAntiviralAntivirals for Hepatitis B agents, physical damage, infectionsInfectionsInvasion of the host organism by microorganisms or their toxins or by parasites that can cause pathological conditions or diseases.Chronic Granulomatous Disease) that also present with hemolysis leading to anemiaAnemiaAnemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types (but not with thrombosisThrombosisFormation and development of a thrombus or blood clot in the blood vessel.Epidemic Typhus and bone marrowBone marrowThe soft tissue filling the cavities of bones. Bone marrow exists in two types, yellow and red. Yellow marrow is found in the large cavities of large bones and consists mostly of fat cells and a few primitive blood cells. Red marrow is a hematopoietic tissue and is the site of production of erythrocytes and granular leukocytes. Bone marrow is made up of a framework of connective tissue containing branching fibers with the frame being filled with marrow cells.Bone Marrow: Composition and Hematopoiesis failure). This is a broad category, as the hemolytic anemias each have a unique mechanism, presentation, and management.
Aplastic anemiaAplastic AnemiaAplastic anemia (AA) is a rare, life-threatening condition characterized by pancytopenia and hypocellularity of the bone marrow (in the absence of any abnormal cells) reflecting damage to hematopoietic stem cells. Aplastic anemia can be acquired or inherited, however, most cases of AA are acquired and caused by autoimmune damage to hematopoietic stem cells. Aplastic Anemia: occurs as a result of hematopoietic stem cell damage. More than 10% of cases will develop into PNH. In addition, some people with PNH will develop aplastic anemiaAplastic AnemiaAplastic anemia (AA) is a rare, life-threatening condition characterized by pancytopenia and hypocellularity of the bone marrow (in the absence of any abnormal cells) reflecting damage to hematopoietic stem cells. Aplastic anemia can be acquired or inherited, however, most cases of AA are acquired and caused by autoimmune damage to hematopoietic stem cells. Aplastic Anemia. Like PNH, aplastic anemiaAplastic AnemiaAplastic anemia (AA) is a rare, life-threatening condition characterized by pancytopenia and hypocellularity of the bone marrow (in the absence of any abnormal cells) reflecting damage to hematopoietic stem cells. Aplastic anemia can be acquired or inherited, however, most cases of AA are acquired and caused by autoimmune damage to hematopoietic stem cells. Aplastic Anemia presents with bone marrowBone marrowThe soft tissue filling the cavities of bones. Bone marrow exists in two types, yellow and red. Yellow marrow is found in the large cavities of large bones and consists mostly of fat cells and a few primitive blood cells. Red marrow is a hematopoietic tissue and is the site of production of erythrocytes and granular leukocytes. Bone marrow is made up of a framework of connective tissue containing branching fibers with the frame being filled with marrow cells.Bone Marrow: Composition and Hematopoiesis failure and low blood cell counts (but not thrombosisThrombosisFormation and development of a thrombus or blood clot in the blood vessel.Epidemic Typhus). Treatment might include medications, blood transfusionsBlood transfusionsThe introduction of whole blood or blood component directly into the bloodstream.Transfusion Products, or stem cell transplantation.
Acute mesenteric ischemiaAcute Mesenteric IschemiaMesenteric Ischemia: presents with severe acute abdominal painAbdominal PainAcute Abdomen in a patient with an identifiable source of thrombus/emboli obstructing the mesenteric vessels (e.g., preexisting atheroma, atrial fibrillationAtrial fibrillationAtrial fibrillation (AF or Afib) is a supraventricular tachyarrhythmia and the most common kind of arrhythmia. It is caused by rapid, uncontrolled atrial contractions and uncoordinated ventricular responses. Atrial Fibrillation). Diagnosed with angiographyAngiographyRadiography of blood vessels after injection of a contrast medium.Cardiac Surgery. Treated with embolectomy or surgery to remove necrotic bowel.
Paroxysmal cold hemoglobinuriaHemoglobinuriaThe presence of free hemoglobin in the urine, indicating hemolysis of erythrocytes within the vascular system. After saturating the hemoglobin-binding proteins (haptoglobins), free hemoglobin begins to appear in the urine.Transfusion Reactions: rare autoimmune hemolytic anemiaAutoimmune Hemolytic AnemiaAutoimmune hemolytic anemia (AIHA) is a rare type of hemolytic anemia characterized by antibody production against self RBCs, leading to destruction of these cells in the spleen and other reticuloendothelial tissues. The disease is generally categorized as warm or cold, depending on the thermal reactivity of the autoantibodies. Autoimmune Hemolytic Anemia seen in young children, triggered by infectious disease, neoplasm, or immune dysfunction. IgMIgMA class of immunoglobulin bearing mu chains (immunoglobulin mu-chains). Igm can fix complement. The name comes from its high molecular weight and originally being called a macroglobulin.Immunoglobulins: Types and FunctionsantibodiesAntibodiesImmunoglobulins (Igs), also known as antibodies, are glycoprotein molecules produced by plasma cells that act in immune responses by recognizing and binding particular antigens. The various Ig classes are IgG (the most abundant), IgM, IgE, IgD, and IgA, which differ in their biologic features, structure, target specificity, and distribution.Immunoglobulins: Types and FunctionsbindBINDHyperbilirubinemia of the Newborn to the RBCsRBCsErythrocytes, or red blood cells (RBCs), are the most abundant cells in the blood. While erythrocytes in the fetus are initially produced in the yolk sac then the liver, the bone marrow eventually becomes the main site of production.Erythrocytes: Histology in the cold and fix complement. Upon warming, the antibodiesAntibodiesImmunoglobulins (Igs), also known as antibodies, are glycoprotein molecules produced by plasma cells that act in immune responses by recognizing and binding particular antigens. The various Ig classes are IgG (the most abundant), IgM, IgE, IgD, and IgA, which differ in their biologic features, structure, target specificity, and distribution.Immunoglobulins: Types and Functions dissociate and complement lyses the RBCsRBCsErythrocytes, or red blood cells (RBCs), are the most abundant cells in the blood. While erythrocytes in the fetus are initially produced in the yolk sac then the liver, the bone marrow eventually becomes the main site of production.Erythrocytes: Histology, leading to extravascular hemolysisExtravascular hemolysisHemolytic Anemia. Diagnosis is via Coombs testing. Treatment is preventive.
Portal veinPortal veinA short thick vein formed by union of the superior mesenteric vein and the splenic vein.Liver: Anatomy obstruction: usually caused by primary thrombosisThrombosisFormation and development of a thrombus or blood clot in the blood vessel.Epidemic Typhus of the portal veinPortal veinA short thick vein formed by union of the superior mesenteric vein and the splenic vein.Liver: Anatomy due to an inherited or acquired coagulopathy or associated with cirrhosisCirrhosisCirrhosis is a late stage of hepatic parenchymal necrosis and scarring (fibrosis) most commonly due to hepatitis C infection and alcoholic liver disease. Patients may present with jaundice, ascites, and hepatosplenomegaly. Cirrhosis can also cause complications such as hepatic encephalopathy, portal hypertension, portal vein thrombosis, and hepatorenal syndrome. Cirrhosis. Extrinsic obstruction also occurs. Correlative clinical findings raise clinical suspicion, but imaging (ultrasound or CT) showing a massMassThree-dimensional lesion that occupies a space within the breastImaging of the Breast or thrombus is diagnostic. Treatment includes thrombectomyThrombectomySurgical removal of an obstructing clot or foreign material from a blood vessel at the point of its formation. Removal of a clot arising from a distant site is called embolectomy.Vascular Surgery, excision of the massMassThree-dimensional lesion that occupies a space within the breastImaging of the Breast/tumorTumorInflammation, and/or anticoagulationAnticoagulationPulmonary Hypertension Drugs.
Renal veinRenal veinShort thick veins which return blood from the kidneys to the vena cava.Glomerular FiltrationthrombosisThrombosisFormation and development of a thrombus or blood clot in the blood vessel.Epidemic Typhus: The most common cause of renal veinRenal veinShort thick veins which return blood from the kidneys to the vena cava.Glomerular FiltrationthrombosisThrombosisFormation and development of a thrombus or blood clot in the blood vessel.Epidemic Typhus is nephrotic syndromeNephrotic syndromeNephrotic syndrome is characterized by severe proteinuria, hypoalbuminemia, and peripheral edema. In contrast, the nephritic syndromes present with hematuria, variable loss of renal function, and hypertension, although there is sometimes overlap of > 1 glomerular disease in the same individual. Nephrotic Syndrome. Other causes include primary hypercoagulableHypercoagulableHypercoagulable states (also referred to as thrombophilias) are a group of hematologic diseases defined by an increased risk of clot formation (i.e., thrombosis) due to either an increase in procoagulants, a decrease in anticoagulants, or a decrease in fibrinolysis. Hypercoagulable States disorders (such as PNH), renal tumors, extrinsic compressionCompressionBlunt Chest Trauma, trauma, and, rarely, inflammatory bowel disease. This condition presents with renal failureRenal failureConditions in which the kidneys perform below the normal level in the ability to remove wastes, concentrate urine, and maintain electrolyte balance; blood pressure; and calcium metabolism. Renal insufficiency can be classified by the degree of kidney damage (as measured by the level of proteinuria) and reduction in glomerular filtration rate.Crush Syndrome (acute or chronic). Diagnosed with imaging (ultrasound or CT). Treatment includes thrombectomyThrombectomySurgical removal of an obstructing clot or foreign material from a blood vessel at the point of its formation. Removal of a clot arising from a distant site is called embolectomy.Vascular Surgery, excision of the massMassThree-dimensional lesion that occupies a space within the breastImaging of the Breast/tumorTumorInflammation, and/or anticoagulationAnticoagulationPulmonary Hypertension Drugs.
References
Luzzatto, L. (2018). Hemolytic anemias. In Jameson, J.L., et al. (Ed.), Harrison’s Principles of Internal Medicine, 20th ed. Vol 1. McGraw Hill. pp. 708–723.
Parker, C., Omine, M., Richards, S., et al. (2005). Diagnosis and management of paroxysmal nocturnal hemoglobinuria. Blood 106:3699–3709. https://doi.org/10.1182/blood-2005-04-1717
Create your free account or log in to continue reading!