Autoimmune hemolytic anemia Hemolytic Anemia Hemolytic anemia (HA) is the term given to a large group of anemias that are caused by the premature destruction/hemolysis of circulating red blood cells (RBCs). Hemolysis can occur within (intravascular hemolysis) or outside the blood vessels (extravascular hemolysis). Hemolytic Anemia (AIHA) is a rare type of hemolytic anemia Hemolytic Anemia Hemolytic anemia (HA) is the term given to a large group of anemias that are caused by the premature destruction/hemolysis of circulating red blood cells (RBCs). Hemolysis can occur within (intravascular hemolysis) or outside the blood vessels (extravascular hemolysis). Hemolytic Anemia characterized by antibody production against self RBCs RBCs Erythrocytes, or red blood cells (RBCs), are the most abundant cells in the blood. While erythrocytes in the fetus are initially produced in the yolk sac then the liver, the bone marrow eventually becomes the main site of production. Erythrocytes: Histology, leading to destruction of these cells in the spleen Spleen The spleen is the largest lymphoid organ in the body, located in the LUQ of the abdomen, superior to the left kidney and posterior to the stomach at the level of the 9th-11th ribs just below the diaphragm. The spleen is highly vascular and acts as an important blood filter, cleansing the blood of pathogens and damaged erythrocytes. Spleen: Anatomy and other reticuloendothelial tissues. The disease is generally categorized as warm or cold, depending on the thermal reactivity of the autoantibodies Autoantibodies Antibodies that react with self-antigens (autoantigens) of the organism that produced them. Blotting Techniques. The etiology is diverse and includes infections Infections Invasion of the host organism by microorganisms or their toxins or by parasites that can cause pathological conditions or diseases. Chronic Granulomatous Disease, autoimmune diseases Autoimmune diseases Disorders that are characterized by the production of antibodies that react with host tissues or immune effector cells that are autoreactive to endogenous peptides. Selective IgA Deficiency, lymphoproliferative disorders Lymphoproliferative disorders Disorders characterized by proliferation of lymphoid tissue, general or unspecified. Lymphocytosis, and drugs. The diagnosis is based on laboratory evidence of hemolysis (elevated LDH LDH Osteosarcoma, indirect bilirubin Indirect Bilirubin Liver Function Tests, reduced haptoglobin) in association with a positive direct antiglobulin test (DAT) or Coombs test. Management involves supportive care, removal of the inciting agent, use of steroids Steroids A group of polycyclic compounds closely related biochemically to terpenes. They include cholesterol, numerous hormones, precursors of certain vitamins, bile acids, alcohols (sterols), and certain natural drugs and poisons. Steroids have a common nucleus, a fused, reduced 17-carbon atom ring system, cyclopentanoperhydrophenanthrene. Most steroids also have two methyl groups and an aliphatic side-chain attached to the nucleus. Benign Liver Tumors and immunosuppressive agents, and splenectomy Splenectomy Surgical procedure involving either partial or entire removal of the spleen. Rupture of the Spleen in refractory cases.
Last updated: Dec 15, 2025
Autoimmune hemolytic anemia Hemolytic Anemia Hemolytic anemia (HA) is the term given to a large group of anemias that are caused by the premature destruction/hemolysis of circulating red blood cells (RBCs). Hemolysis can occur within (intravascular hemolysis) or outside the blood vessels (extravascular hemolysis). Hemolytic Anemia (AIHA) is hemolysis or premature Premature Childbirth before 37 weeks of pregnancy (259 days from the first day of the mother’s last menstrual period, or 245 days after fertilization). Necrotizing Enterocolitis destruction of RBCs RBCs Erythrocytes, or red blood cells (RBCs), are the most abundant cells in the blood. While erythrocytes in the fetus are initially produced in the yolk sac then the liver, the bone marrow eventually becomes the main site of production. Erythrocytes: Histology due to autoantibodies Autoantibodies Antibodies that react with self-antigens (autoantigens) of the organism that produced them. Blotting Techniques. There are 2 categories of AIHA based on the thermal reactivity of autoantibodies Autoantibodies Antibodies that react with self-antigens (autoantigens) of the organism that produced them. Blotting Techniques:
Warm AIHA:
Cold AIHA:
Mixed-type AIHA:
| Type | Specific cause | |
|---|---|---|
| Primary | Idiopathic Idiopathic Dermatomyositis | |
| Secondary | Viral infection | HIV HIV Anti-HIV Drugs |
| Other disease/condition |
|
|
| Drugs |
|
|
| Type | Specific cause | |
|---|---|---|
| Primary | Primary cold agglutinin disease (CAD) | |
| Secondary | Viral infection |
|
| Bacterial infection |
|
|
| Other conditions |
|
|
| Drug | Lenalidomide Lenalidomide Has immunomodulatory (↓ tumor necrosis factor-⍺, ↑ natural killer cells and IL-2) and antiangiogenic activity. Induces cell-cycle arrest and death → inhibiting tumor cell growth. Cancer Immunotherapy (rare) | |
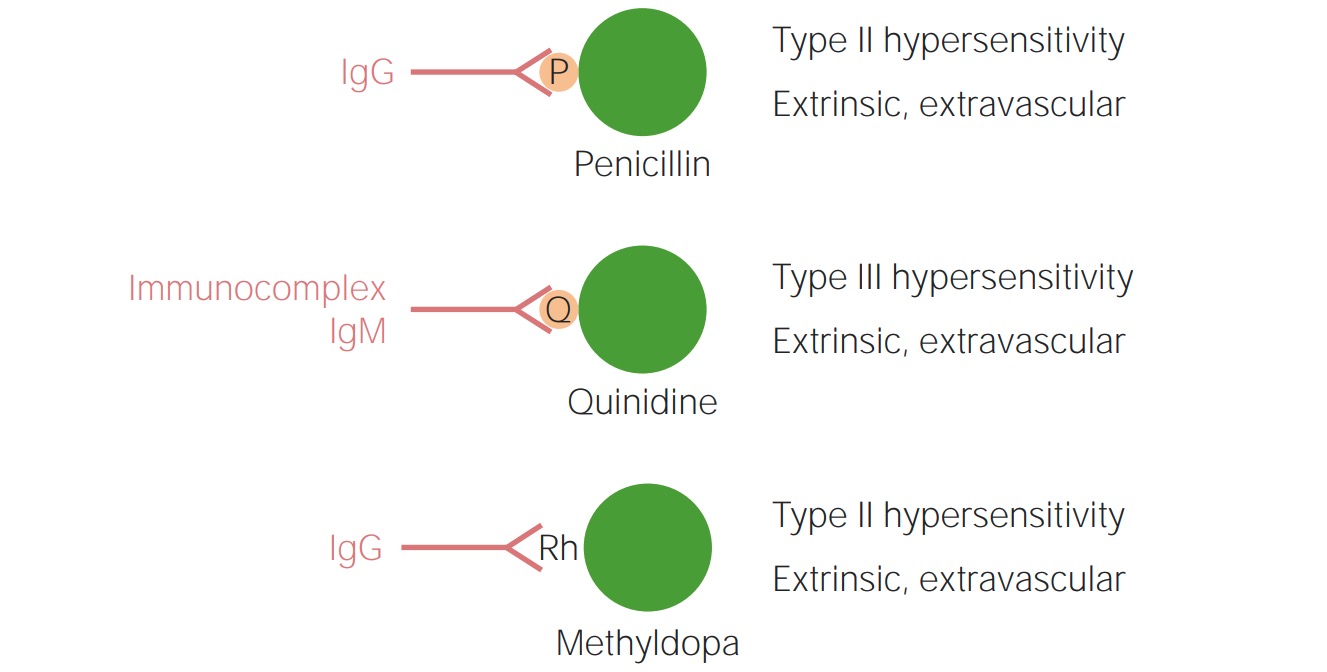
Drug-induced autoimmune hemolytic anemia (AIHA):
Drugs can cause AIHA by:
Top: Binding to specific proteins of a drug that remain on the surface of RBCs and are recognized as new antigens by IgG (penicillin)
Middle: Complement activation induced by immune complexes formed by a drug (quinidine)
Bottom: Alteration of the Rhesus factor (Rh) molecule or a cell membrane component by a drug, thus making it immunogenic for IgG even after the drug is discontinued (methyldopa)
Cold-sensitive autoantibodies Autoantibodies Antibodies that react with self-antigens (autoantigens) of the organism that produced them. Blotting Techniques react more strongly at 0–4℃.
Cold AIHA:
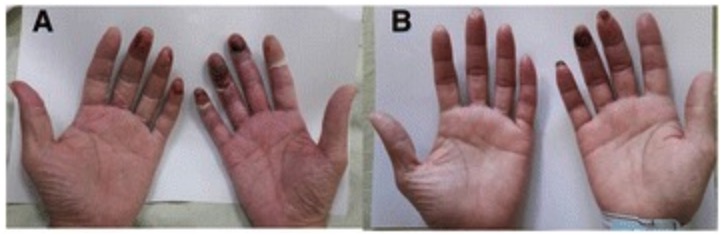
Acrocyanosis of the hands:
Acrocyanosis with gangrene (A) and resolution of acrocyanosis (B)
Diagnosis is with evidence of hemolysis and positive DAT.
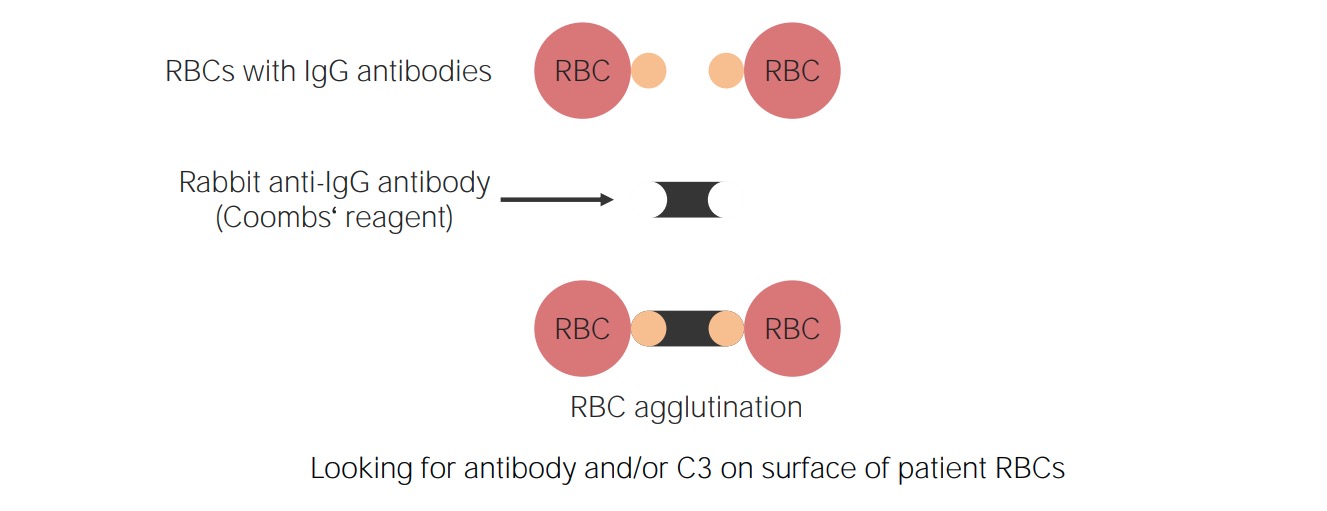
Principle of direct Coombs test:
Antihuman immunoglobulin reagent (directed against immunoglobulins, specifically IgG, and a fragment of the 3rd component of complement (C3d)) are added to the blood sample. Reagent antibodies will bind and agglutinate the RBCs if they already have surface IgG and/or C3d.
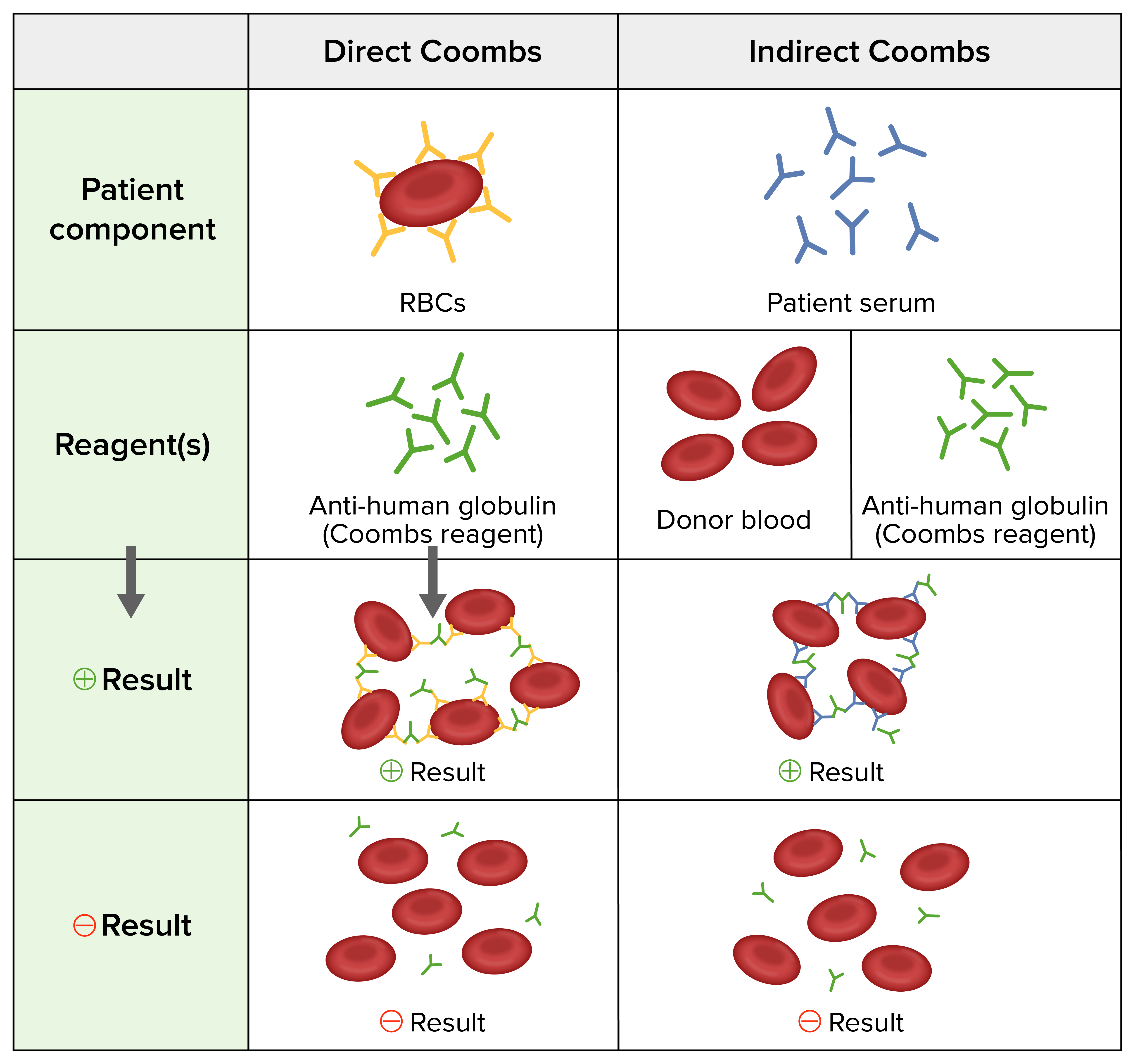
Direct and indirect Coombs tests:
Direct Coombs test: Anti-human antibodies are added to the individual’s washed RBCs.
Indirect Coombs test: Donor RBCs and anti-human antibodies are added to the individual’s serum.
Positive result: agglutination of RBCs
Negative result: no agglutination of RBCs
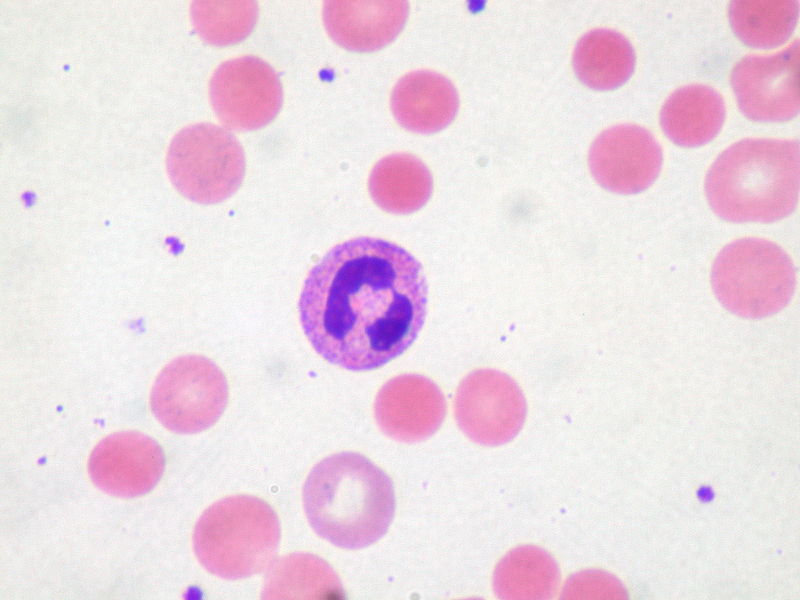
Peripheral blood smear:
Spherocytes are small round RBCs caused by partial phagocytosis of their membrane by spleen phagocytes (induced by membrane-bound antigens).
Warm AIHA:
Cold AIHA: