Chronic lymphocytic leukemia (CLL) is a hematologic malignancy Malignancy Hemothorax characterized by excess production of fully differentiated monoclonal B lymphocytes B lymphocytes Lymphoid cells concerned with humoral immunity. They are short-lived cells resembling bursa-derived lymphocytes of birds in their production of immunoglobulin upon appropriate stimulation. B cells: Types and Functions in the peripheral blood. When the involvement is primarily nodal, the condition is called small lymphocytic lymphoma Lymphoma A general term for various neoplastic diseases of the lymphoid tissue. Imaging of the Mediastinum (SLL). The disease usually presents in older adults, with a median age of 70 years. Clinical presentation is often asymptomatic and diagnosis is made when abnormal lymphocytosis Lymphocytosis WBCs develop from stem cells in the bone marrow and are called leukocytes when circulating in the bloodstream. Lymphocytes are 1 of the 5 subclasses of WBCs. Lymphocytosis is an increase in the number or proportion of the lymphocyte subclass of WBCs, often as a result of an immune response to infection (known as reactive lymphocytosis). Lymphocytosis is seen on laboratory testing. Cytopenias Cytopenias IPEX Syndrome can also be seen. The B cells B cells Lymphoid cells concerned with humoral immunity. They are short-lived cells resembling bursa-derived lymphocytes of birds in their production of immunoglobulin upon appropriate stimulation. B cells: Types and Functions are functionally incompetent lymphocytes Lymphocytes Lymphocytes are heterogeneous WBCs involved in immune response. Lymphocytes develop from the bone marrow, starting from hematopoietic stem cells (HSCs) and progressing to common lymphoid progenitors (CLPs). B and T lymphocytes and natural killer (NK) cells arise from the lineage. Lymphocytes: Histology; thus, recurrent infections Recurrent infections Common Variable Immunodeficiency (CVID) can sometimes be seen with this malignancy Malignancy Hemothorax. Treatment can vary from watchful waiting for early stages to chemotherapy Chemotherapy Osteosarcoma for later stages. Median survival is around 10 years.
Last updated: Dec 15, 2025
Chronic lymphocytic leukemia (CLL) is a lymphoproliferative neoplasm characterized by accumulation of functionally impaired lymphocytes Lymphocytes Lymphocytes are heterogeneous WBCs involved in immune response. Lymphocytes develop from the bone marrow, starting from hematopoietic stem cells (HSCs) and progressing to common lymphoid progenitors (CLPs). B and T lymphocytes and natural killer (NK) cells arise from the lineage. Lymphocytes: Histology, often monoclonal in origin.
CLL is identical to small lymphocytic lymphoma Lymphoma A general term for various neoplastic diseases of the lymphoid tissue. Imaging of the Mediastinum (SLL), both of which are considered manifestations of B-cell neoplasm.
Creation of new blood cells ( hematopoiesis Hematopoiesis The development and formation of various types of blood cells. Hematopoiesis can take place in the bone marrow (medullary) or outside the bone marrow (extramedullary hematopoiesis). Bone Marrow: Composition and Hematopoiesis) starts with a hematopoietic stem cell, which is prompted to divide and differentiate with appropriate chemical stimuli (hemopoietic growth factors).
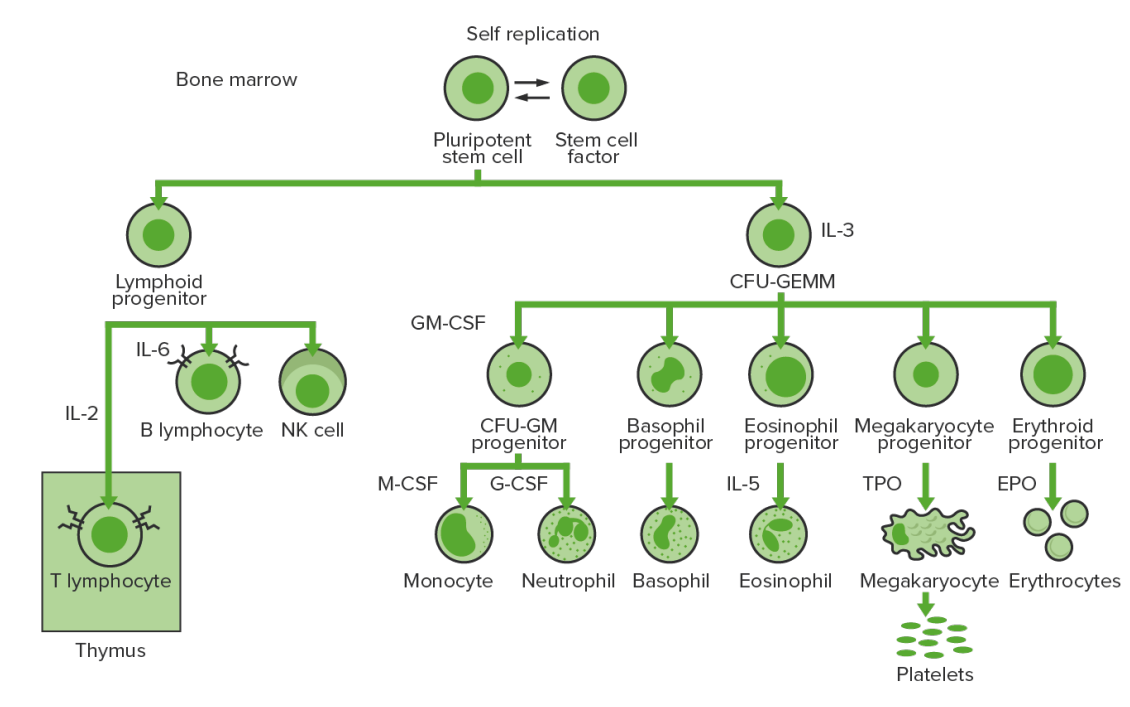
Bone marrow hematopoiesis: proliferation and differentiation of the formed elements of blood.
In CML, there is sustained proliferation of cells in the granulocytic line (myeloblasts → neutrophils, basophils, eosinophils). Both mature and maturing cells are seen; thus, there are cells that are only partially effective in immune response.
IL-3: interleukin-3
CFU-GEMM: colony-forming unit–granulocyte, erythrocyte, monocyte, megakaryocyte
IL-2: interleukin-2
IL-6: interleukin-6
CFU-GM: colony-forming unit–granulocyte-macrophage
GM-CSF: granulocyte-macrophage colony-stimulating factor
M-CSF: macrophage colony-stimulating factor
G-CSF: granulocyte colony-stimulating factor
IL-5: interleukin-5
NK: natural killer
TPO: thrombopoietin
EPO: erythropoietin
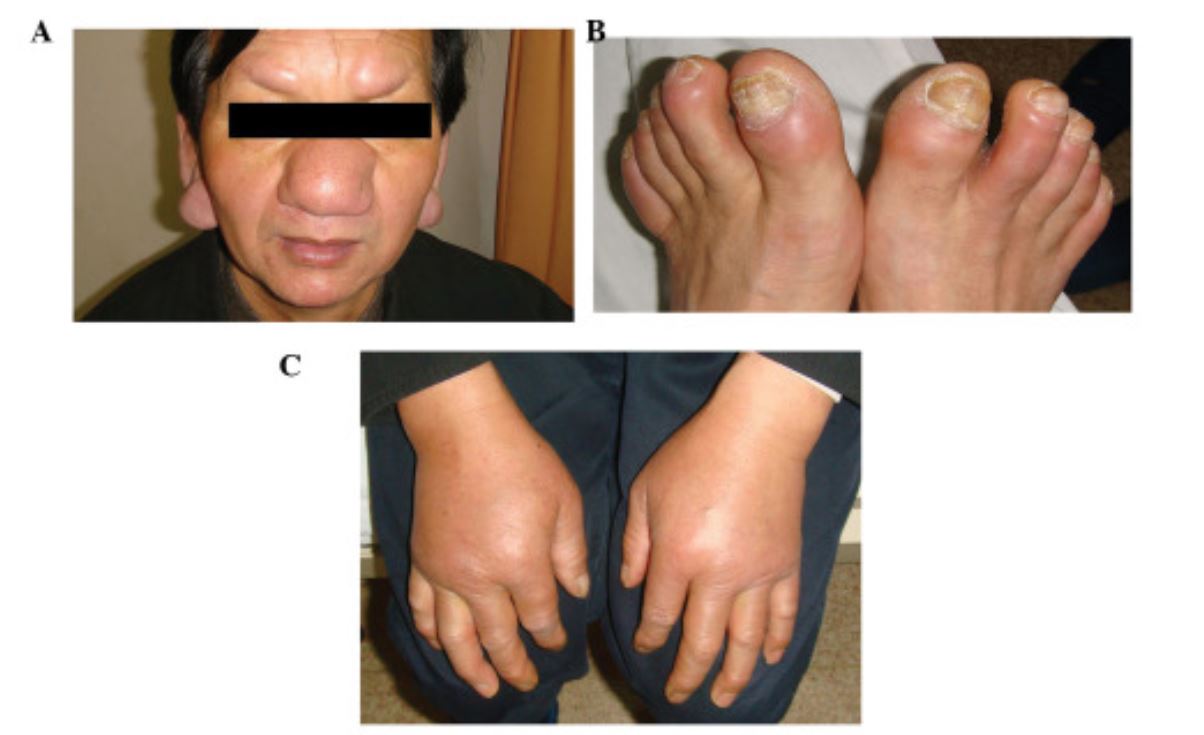
Rare CLL infiltration of the ears, eyebrows, and nose (A) and the toes (B). Bilateral nonpitting edema on the backs of the hands (C).
Image: “Cutaneous manifestations in a patient with chronic lymphocytic leukemia involving the head, neck and distal extremities” by Lu C, Li L, Qiao Q, Liu G, Fang L. License: CC BY 3.0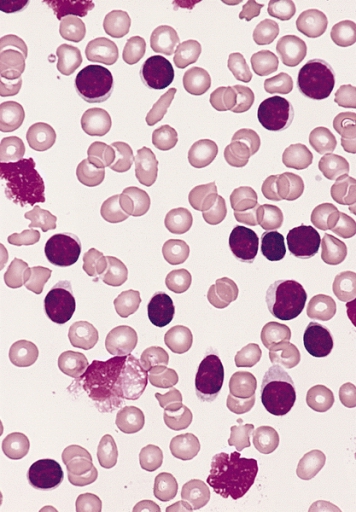
Blood smear from an adult man with marked lymphocytosis:
The predominant lymphocytes have very sparse pale cytoplasm, round to slightly oval nuclei, and no evident nucleoli, and 3 damaged “smudge cells” are present, characteristic of the majority of cases of CLL.
Based on the involvement of 5 possible lymphoid-bearing areas ( lymph Lymph The interstitial fluid that is in the lymphatic system. Secondary Lymphatic Organs node enlargement is ≥ 1 cm):
| Stage | Description | Survival |
|---|---|---|
| A (low risk) | ≤ 2 lymphoid areas enlarged | Same as age-matched controls |
| B (intermediate risk) | ≥ 3 lymphoid areas enlarged | 84 months |
| C (high risk) | Presence of anemia Anemia Anemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types ( Hb Hb The oxygen-carrying proteins of erythrocytes. They are found in all vertebrates and some invertebrates. The number of globin subunits in the hemoglobin quaternary structure differs between species. Structures range from monomeric to a variety of multimeric arrangements. Gas Exchange < 10 g/dL) or thrombocytopenia Thrombocytopenia Thrombocytopenia occurs when the platelet count is < 150,000 per microliter. The normal range for platelets is usually 150,000-450,000/µL of whole blood. Thrombocytopenia can be a result of decreased production, increased destruction, or splenic sequestration of platelets. Patients are often asymptomatic until platelet counts are < 50,000/µL. Thrombocytopenia ( platelets Platelets Platelets are small cell fragments involved in hemostasis. Thrombopoiesis takes place primarily in the bone marrow through a series of cell differentiation and is influenced by several cytokines. Platelets are formed after fragmentation of the megakaryocyte cytoplasm. Platelets: Histology < 100,000/µL) | 24 months |
Classifying 3 different stages, based on:
| Risk | Stage | Description | Survival |
|---|---|---|---|
| Low | 0 | Lymphocytosis Lymphocytosis WBCs develop from stem cells in the bone marrow and are called leukocytes when circulating in the bloodstream. Lymphocytes are 1 of the 5 subclasses of WBCs. Lymphocytosis is an increase in the number or proportion of the lymphocyte subclass of WBCs, often as a result of an immune response to infection (known as reactive lymphocytosis). Lymphocytosis in blood or bone marrow Bone marrow The soft tissue filling the cavities of bones. Bone marrow exists in two types, yellow and red. Yellow marrow is found in the large cavities of large bones and consists mostly of fat cells and a few primitive blood cells. Red marrow is a hematopoietic tissue and is the site of production of erythrocytes and granular leukocytes. Bone marrow is made up of a framework of connective tissue containing branching fibers with the frame being filled with marrow cells. Bone Marrow: Composition and Hematopoiesis | > 150 months |
| Intermediate | I | Lymphocytosis Lymphocytosis WBCs develop from stem cells in the bone marrow and are called leukocytes when circulating in the bloodstream. Lymphocytes are 1 of the 5 subclasses of WBCs. Lymphocytosis is an increase in the number or proportion of the lymphocyte subclass of WBCs, often as a result of an immune response to infection (known as reactive lymphocytosis). Lymphocytosis + enlarged lymph nodes Lymph Nodes They are oval or bean shaped bodies (1 – 30 mm in diameter) located along the lymphatic system. Lymphatic Drainage System: Anatomy | 101 months |
| II | Lymphocytosis Lymphocytosis WBCs develop from stem cells in the bone marrow and are called leukocytes when circulating in the bloodstream. Lymphocytes are 1 of the 5 subclasses of WBCs. Lymphocytosis is an increase in the number or proportion of the lymphocyte subclass of WBCs, often as a result of an immune response to infection (known as reactive lymphocytosis). Lymphocytosis + enlarged liver Liver The liver is the largest gland in the human body. The liver is found in the superior right quadrant of the abdomen and weighs approximately 1.5 kilograms. Its main functions are detoxification, metabolism, nutrient storage (e.g., iron and vitamins), synthesis of coagulation factors, formation of bile, filtration, and storage of blood. Liver: Anatomy or spleen Spleen The spleen is the largest lymphoid organ in the body, located in the LUQ of the abdomen, superior to the left kidney and posterior to the stomach at the level of the 9th-11th ribs just below the diaphragm. The spleen is highly vascular and acts as an important blood filter, cleansing the blood of pathogens and damaged erythrocytes. Spleen: Anatomy with or without lymphadenopathy Lymphadenopathy Lymphadenopathy is lymph node enlargement (> 1 cm) and is benign and self-limited in most patients. Etiologies include malignancy, infection, and autoimmune disorders, as well as iatrogenic causes such as the use of certain medications. Generalized lymphadenopathy often indicates underlying systemic disease. Lymphadenopathy | 71 months | |
| High | III | Lymphocytosis Lymphocytosis WBCs develop from stem cells in the bone marrow and are called leukocytes when circulating in the bloodstream. Lymphocytes are 1 of the 5 subclasses of WBCs. Lymphocytosis is an increase in the number or proportion of the lymphocyte subclass of WBCs, often as a result of an immune response to infection (known as reactive lymphocytosis). Lymphocytosis + anemia Anemia Anemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types (hemoglobin < 11 g/dL) with or without enlarged liver Liver The liver is the largest gland in the human body. The liver is found in the superior right quadrant of the abdomen and weighs approximately 1.5 kilograms. Its main functions are detoxification, metabolism, nutrient storage (e.g., iron and vitamins), synthesis of coagulation factors, formation of bile, filtration, and storage of blood. Liver: Anatomy, spleen Spleen The spleen is the largest lymphoid organ in the body, located in the LUQ of the abdomen, superior to the left kidney and posterior to the stomach at the level of the 9th-11th ribs just below the diaphragm. The spleen is highly vascular and acts as an important blood filter, cleansing the blood of pathogens and damaged erythrocytes. Spleen: Anatomy, or lymph nodes Lymph Nodes They are oval or bean shaped bodies (1 – 30 mm in diameter) located along the lymphatic system. Lymphatic Drainage System: Anatomy | 19 months |
| IV | Lymphocytosis Lymphocytosis WBCs develop from stem cells in the bone marrow and are called leukocytes when circulating in the bloodstream. Lymphocytes are 1 of the 5 subclasses of WBCs. Lymphocytosis is an increase in the number or proportion of the lymphocyte subclass of WBCs, often as a result of an immune response to infection (known as reactive lymphocytosis). Lymphocytosis + thrombocytopenia Thrombocytopenia Thrombocytopenia occurs when the platelet count is < 150,000 per microliter. The normal range for platelets is usually 150,000-450,000/µL of whole blood. Thrombocytopenia can be a result of decreased production, increased destruction, or splenic sequestration of platelets. Patients are often asymptomatic until platelet counts are < 50,000/µL. Thrombocytopenia (platelet count < 100,000/µL) with or without anemia Anemia Anemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types or enlarged liver Liver The liver is the largest gland in the human body. The liver is found in the superior right quadrant of the abdomen and weighs approximately 1.5 kilograms. Its main functions are detoxification, metabolism, nutrient storage (e.g., iron and vitamins), synthesis of coagulation factors, formation of bile, filtration, and storage of blood. Liver: Anatomy, spleen Spleen The spleen is the largest lymphoid organ in the body, located in the LUQ of the abdomen, superior to the left kidney and posterior to the stomach at the level of the 9th-11th ribs just below the diaphragm. The spleen is highly vascular and acts as an important blood filter, cleansing the blood of pathogens and damaged erythrocytes. Spleen: Anatomy, or lymph nodes Lymph Nodes They are oval or bean shaped bodies (1 – 30 mm in diameter) located along the lymphatic system. Lymphatic Drainage System: Anatomy |