


A leucemia linfocítica crónica (LLC) é uma doença hematológica maligna caracterizada pela produção excessiva de linfócitos B monoclonais completamente diferenciados no sangue periférico. Quando o envolvimento é maioritariamente ganglionar, a doença é denominada linfoma linfocítico de pequenas células (LLPC). Apresenta-se geralmente em idosos, com uma mediana de idades de 70 anos. Os doentes são geralmente assintomáticos e o diagnóstico é realizado pela deteção de linfocitose anormal em análises laboratoriais. Podem também ocorrer citopenias. As células B são linfócitos funcionalmente incompetentes; assim por vezes pode ver-se em infeções recorrentes com esta neoplasia. O tratamento pode variar desde a observação (watchful waiting) para os estadios iniciais até quimioterapia para os estadios mais MAIS Androgen Insensitivity Syndrome avançados. A sobrevida média é de cerca de 10 anos.
Last updated: Dec 15, 2025
A leucemia linfocítica crónica (LLC) é uma neoplasia linfoproliferativa caracterizada pela acumulação de linfócitos funcionalmente incompetentes, geralmente com origem monoclonal.
A LLC é semelhante ao linfoma linfocítico de pequenas células (LLPC), sendo que ambos são considerados manifestações de neoplasia de células B.
A produção de novas células sanguíneas (hematopoiese) inicia-se com uma célula estaminal hematopoiética, estimulada a dividir-se e a diferenciar-se com estímulos químicos apropriados (fatores de crescimento hematopoiéticos).
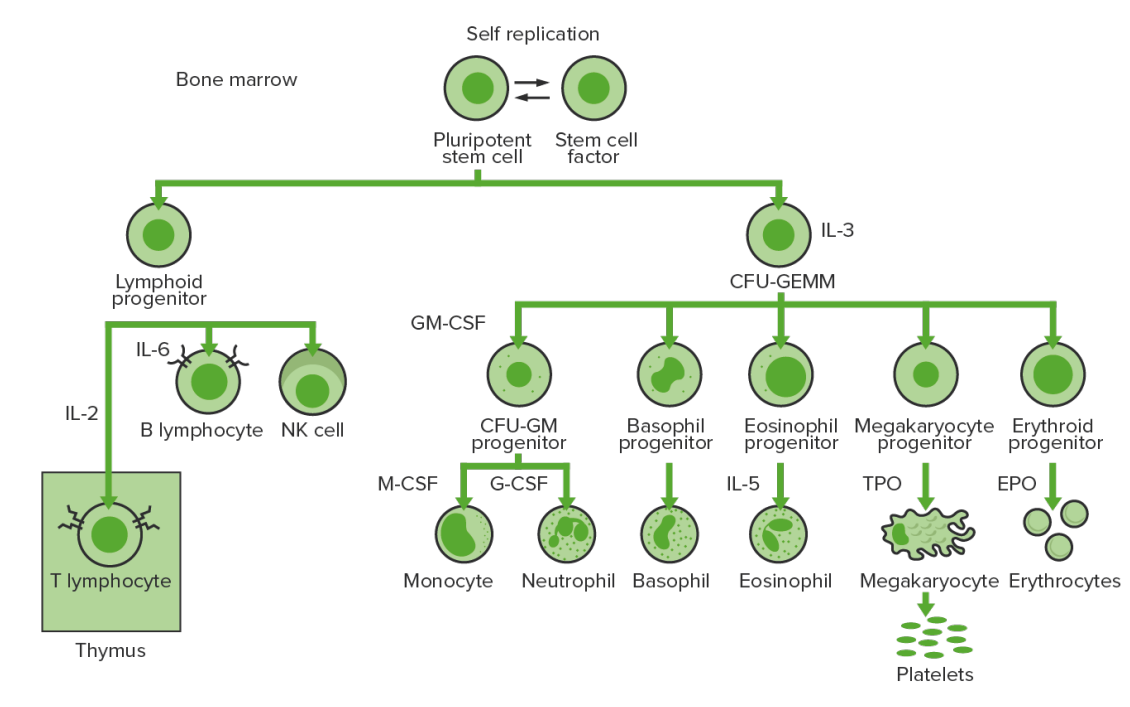
Hematopoiese da medula óssea:
Proliferação e diferenciação de elementos sanguíneos:
Na LMC, ocorre proliferação sustentada de células da linhagem granulocítica (mieloblastos → neutrófilos, basófilos, eosinófilos). É possível observar células maduras e em maturação; assim, existem células que são apenas parcialmente eficazes na resposta imune.
CFU-GEMM (pela sigla em inglês): unidade formadora de colónias de granulócitos, eritrócitos, monócitos, megacariócitos
CFU-GM (pela sigla em inglês): unidade formadora de colónias de macrófagos e granulócitos
GM-CSF (pela sigla em inglês): fator estimulador de colónias de granulócitos e macrófagos
M-CSF (pela sigla em inglês): fator estimulador de colónias de macrófagos
G-CSF (pela sigla em inglês): fator estimulador de colónias de granulócitos
NK: natural killer
TPO: trombopoietina
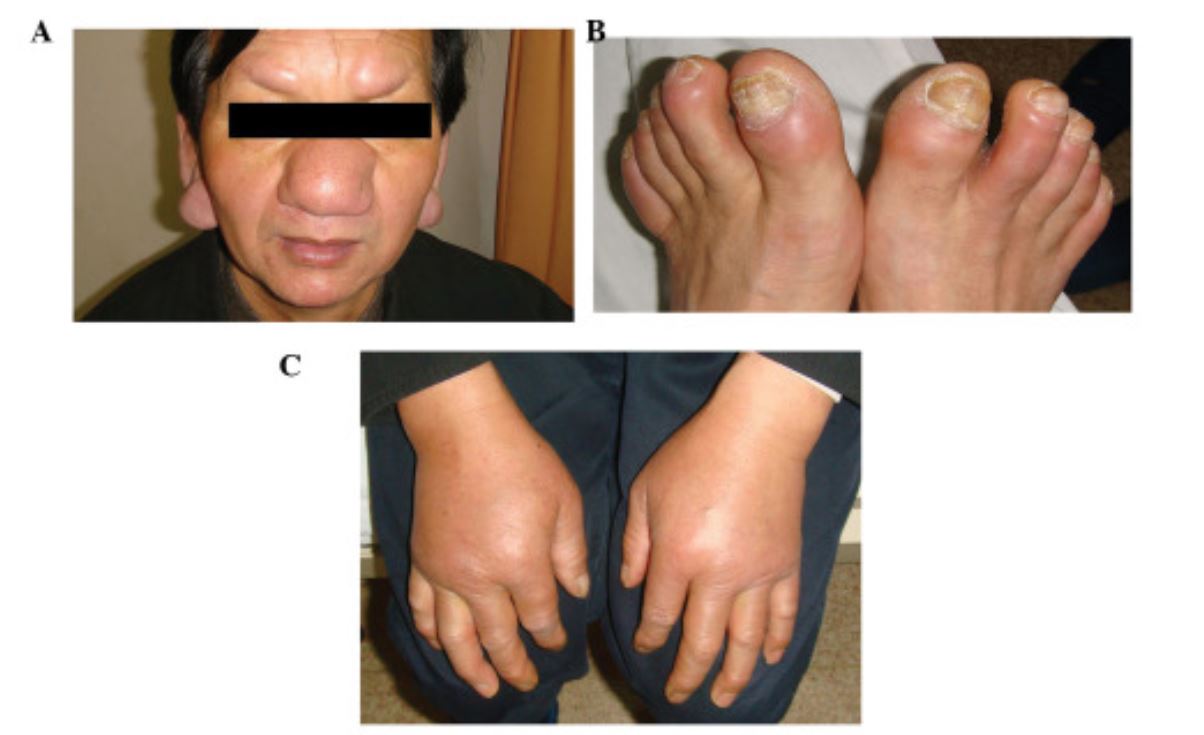
Infiltração rara de LLC nas orelhas, sobrancelhas, nariz (A) e dedos dos pés (B). Edema bilateral não depressível no dorso das mãos (C).
Imagem: “Cutaneous manifestations in a patient with chronic lymphocytic leukemia involving the head, neck and distal extremities” por Lu C, Li L, Qiao Q, Liu G, Fang L. License: CC BY 3.0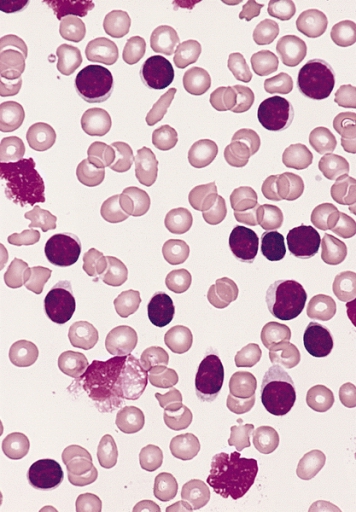
Esfregaço de sangue periférico de um indivíduo adulto do sexo masculino com linfocitose marcada:
Os linfócitos predominantes apresentam um citoplasma pálido muito disperso, núcleos redondos/ligeiramente ovais, sem evidência de nucléolo e 3 smudge cells, características da maioria dos casos de LLC.
Com base no envolvimento de 5 possíveis áreas linfóides (considera-se aumento do tamanho de gânglios linfáticos ≥ 1 cm):
| Estadio | Descrição | Sobrevivência |
|---|---|---|
| A (baixo risco) | ≤ 2 áreas linfóides aumentadas | Igual aos controlos em doentes com a mesma idade |
| B (risco intermédio) | ≥ 3 áreas linfóides aumentadas | 84 meses |
| C (alto risco) | Presença de anemia Anemia Anemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types ( Hb Hb The oxygen-carrying proteins of erythrocytes. They are found in all vertebrates and some invertebrates. The number of globin subunits in the hemoglobin quaternary structure differs between species. Structures range from monomeric to a variety of multimeric arrangements. Gas Exchange < 10 g/dL) ou trombocitopenia (plaquetas < 100.000/µL) | 24 meses |
Classificação em 3 estadios distintos, com base em:
| Risco | Estadio | Descrição | Sobrevivência |
|---|---|---|---|
| Baixo | 0 | Linfocitose no sangue ou medula óssea | > 150 meses |
| Intermédio | I | Linfocitose + aumento de gânglios linfáticos | 101 meses |
| II | Linfocitose + hepatomegalia + esplenomegalia com ou sem linfadenopatia | 71 meses | |
| Alto | III | Linfocitose + anemia Anemia Anemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types (hemoglobina < 11 g/dL) com ou sem hepatomegalia, esplenomegalia ou linfadenopatia | 19 meses |
| IV | Linfocitose + trombocitopenia (contagem de plaquetas < 100.000/µL) com ou sem anemia Anemia Anemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types ou hepatomegalia, esplenomegalia ou linfadenopatia |
