


A hemoglobinúria paroxística noturna (HPN) é uma anemia Anemia Anemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types hemolítica adquirida rara, mas grave, com períodos de exacerbação. Esta anemia Anemia Anemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types é causada pela expansão clonal não maligna de ≥ 1 células-estaminais hematopoiéticas que adquiriram uma mutação somática do gene Gene A category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms. Basic Terms of Genetics da subunidade A da fosfatidilinositol N -acetilglucosaminiltransferase (PIG-A). A expansão clonal destas células é deficiente em proteínas ancoradas por glicosilfosfatidilinositol (GPI-APs). A deficiência dos GPI-APs CD55 e CD59, que regulam o complemento, resulta em hemólise intravascular. A tríade clássica da HPN é definida por anemia Anemia Anemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types hemolítica, insuficiência medular e trombofilia. Os pacientes podem ser tratados com o anticorpo monoclonal eculizumab ou com transplante de células-estaminais, além de geridas as complicações associadas.
Last updated: Dec 15, 2025
Hemoglobinúria paroxística noturna clássica (HPN):
HPN no contexto de um distúrbio específico da medula óssea:
HPN subclínica (HPN-sc):
Gene Gene A category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms. Basic Terms of Genetics PIG-A (fisiologia normal) :
Gene Gene A category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms. Basic Terms of Genetics PIG-A (mutação) :
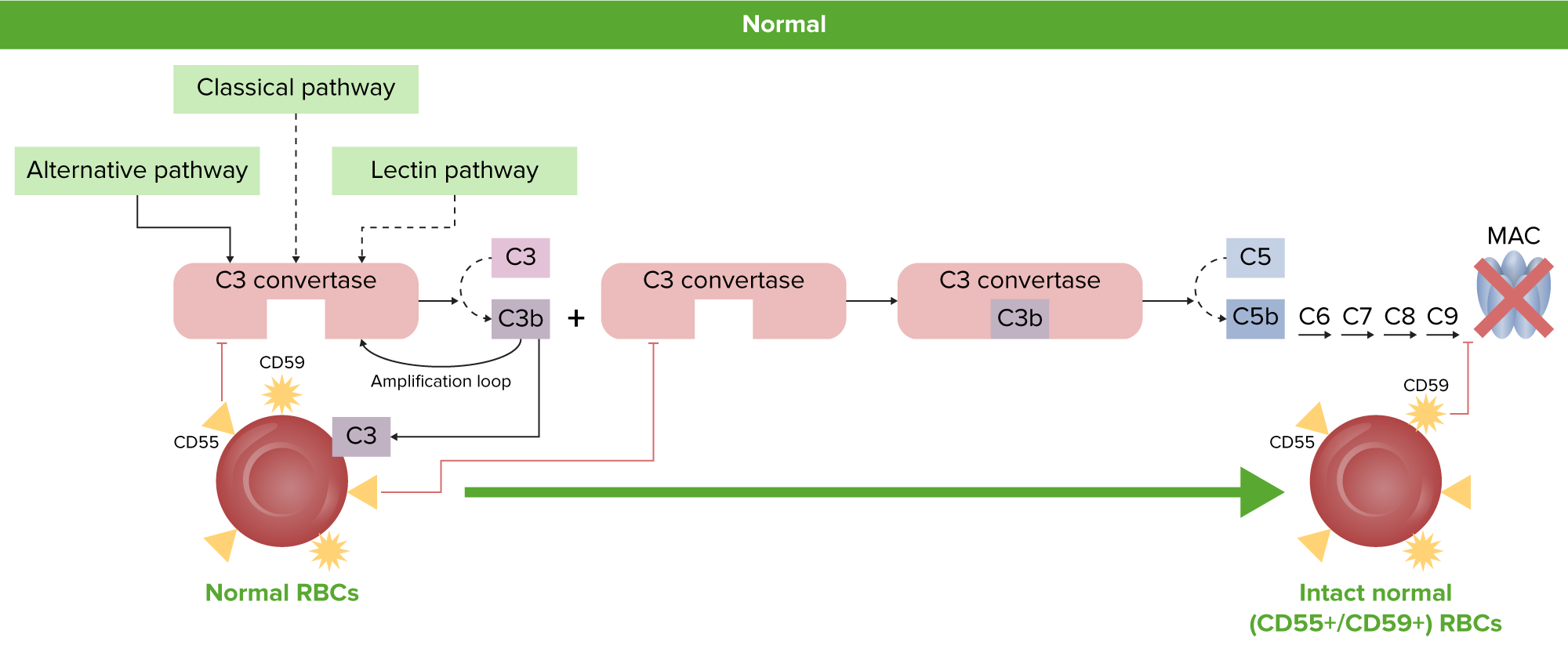
Fisiologia do GV normal:
Os GV normais são protegidas da ativação do complemento e hemólise pelas proteínas reguladoras do complemento ligadas a GPI, CD55 e CD59.
GPI: glicosilfosfatidilinositol
CAM: complexo de ataque à membrana
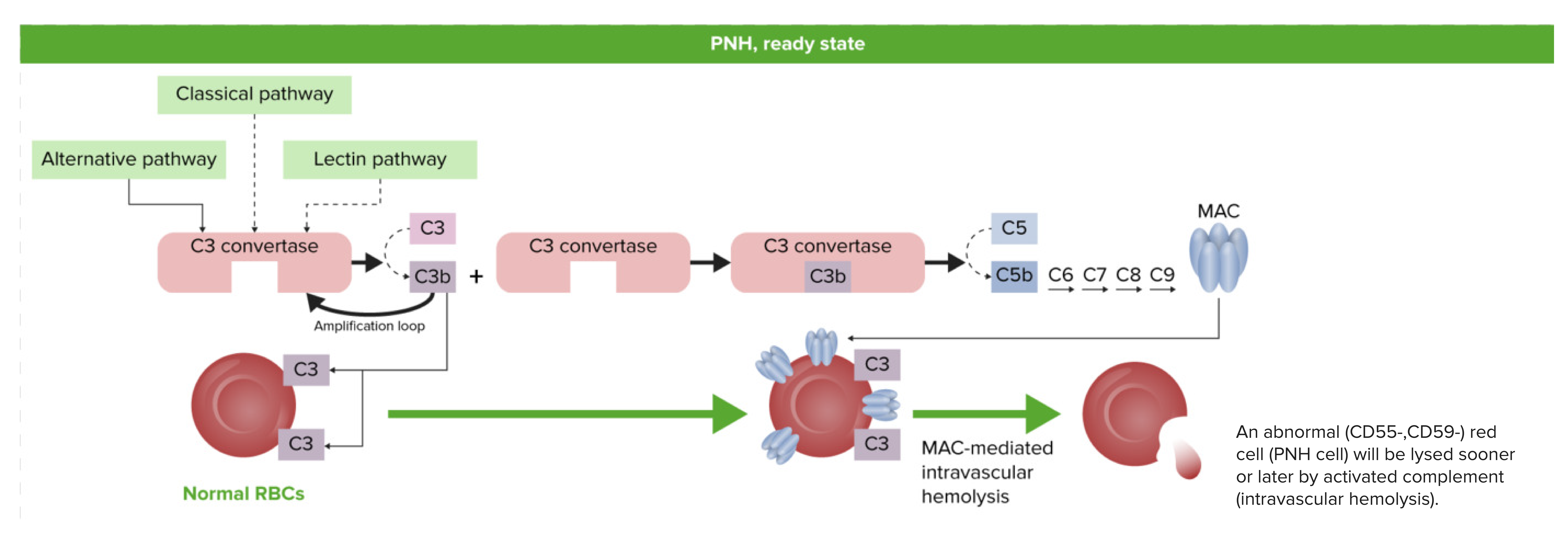
Fisiologia da hemoglobinúria paroxística noturna (HPN) no estado estacionário:
Mesmo na ausência de inflamação, infeção ou outro trigger, os eritrócitos com GPI reduzido ou nenhum na sua superfície são suscetíveis ao ataque do complemento pela ativação espontânea do complemento a partir de uma quantidade mínima, mas constante de C3 ativado (o “tickover C3”). A hemólise intravascular ocorre devido à formação de um complexo de ataque à membrana (CAM), que causa distorção focal e buracos na membrana celular. Se o complemento extra for ativado pela via clássica antígeno-anticorpo (por exemplo, por infeção), então a hemólise será aumentada; isto corresponde à exacerbação paroxística da doença.
GPI: glicosilfosfatidilinositol

Amostra de urina de um paciente com HPN (esquerda) e de um controlo saudável (direita).
A urina do paciente está vermelha devido à hemólise intravascular com hemoglobinúria. A hemólise ocorre ao longo do dia, não apenas à noite.
Os neutrófilos também apresentam deficiência de GPI-APs:
Os fatores mediados por plaquetas incluem:
A trombose em locais incomuns (veias hepáticas, mesentéricas, cerebrais, dérmicas) é típica da HPN.
A insuficiência renal é progressiva e multifatorial.
As exacerbações da hemólise podem ser devidas a:
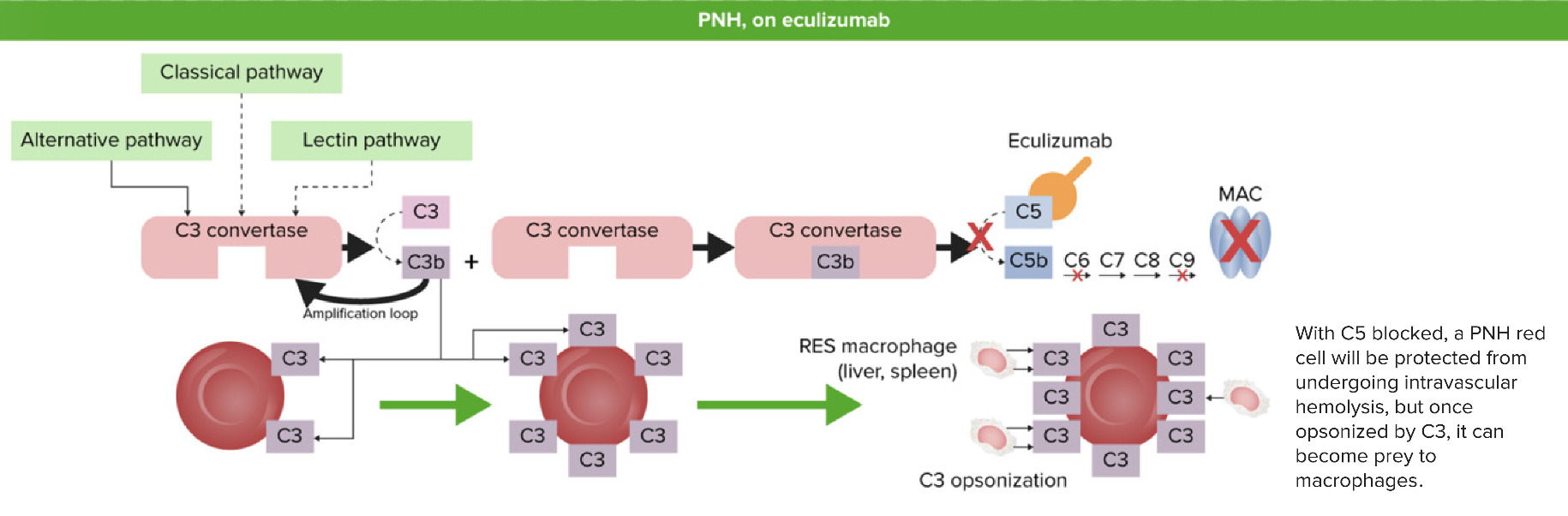
Hemoglobinúria paroxística noturna após eculizumab:
Quando C5 é bloqueado (inibido) pelo eculizumab, a C5 convertase não pode formar C5b a partir da clivagem de C5. O complexo de ataque à membrana (CAM) não é formado e não ocorre hemólise intravascular. No entanto, os fragmentos C3 que foram libertados no início do processo de ativação irão revestir as hemácias desprotegidas e causar hemólise extravascular pelo sistema reticuloendotelial (SRE) no fígado e no baço.
Esses sintomas são devidos à diminuição do ON circulante:
