La hemoglobinuria Hemoglobinuria The presence of free hemoglobin in the urine, indicating hemolysis of erythrocytes within the vascular system. After saturating the hemoglobin-binding proteins (haptoglobins), free hemoglobin begins to appear in the urine. Transfusion Reactions paroxística nocturna (HPN) es una anemia Anemia Anemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types hemolítica adquirida, rara, pero grave, con exacerbaciones periódicas. Esta anemia Anemia Anemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types es causada por una expansión clonal no maligna de ≥ 1 células madre hematopoyéticas que han adquirido una mutación somática del gen de la subunidad A de la fosfatidilinositol N-acetilglucosaminiltransferasa (PIG-A). La expansión clonal de las células madre afectadas son deficientes en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum proteínas ancladas a glicosilfosfatidilinositol. La deficiencia de las proteínas ancladas a glucosilfosfatidilinositol, CD55 y CD59, que regulan el complemento, provocan hemólisis intravascular. La tríada clásica de la HPN es anemia Anemia Anemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types hemolítica, insuficiencia medular y trombofilia. Los LOS Neisseria pacientes pueden ser tratados con el anticuerpo monoclonal eculizumab o con un trasplante de células madre, además de ser tratados por las complicaciones asociadas.
Last updated: Dec 15, 2025
Hemoglobinuria Hemoglobinuria The presence of free hemoglobin in the urine, indicating hemolysis of erythrocytes within the vascular system. After saturating the hemoglobin-binding proteins (haptoglobins), free hemoglobin begins to appear in the urine. Transfusion Reactions paroxística nocturna (HPN) clásica:
HPN en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum el marco de un trastorno específico de la médula ósea:
HPN subclínica:
Gen PIG-A (fisiología normal):
Gen PIG-A (mutación):
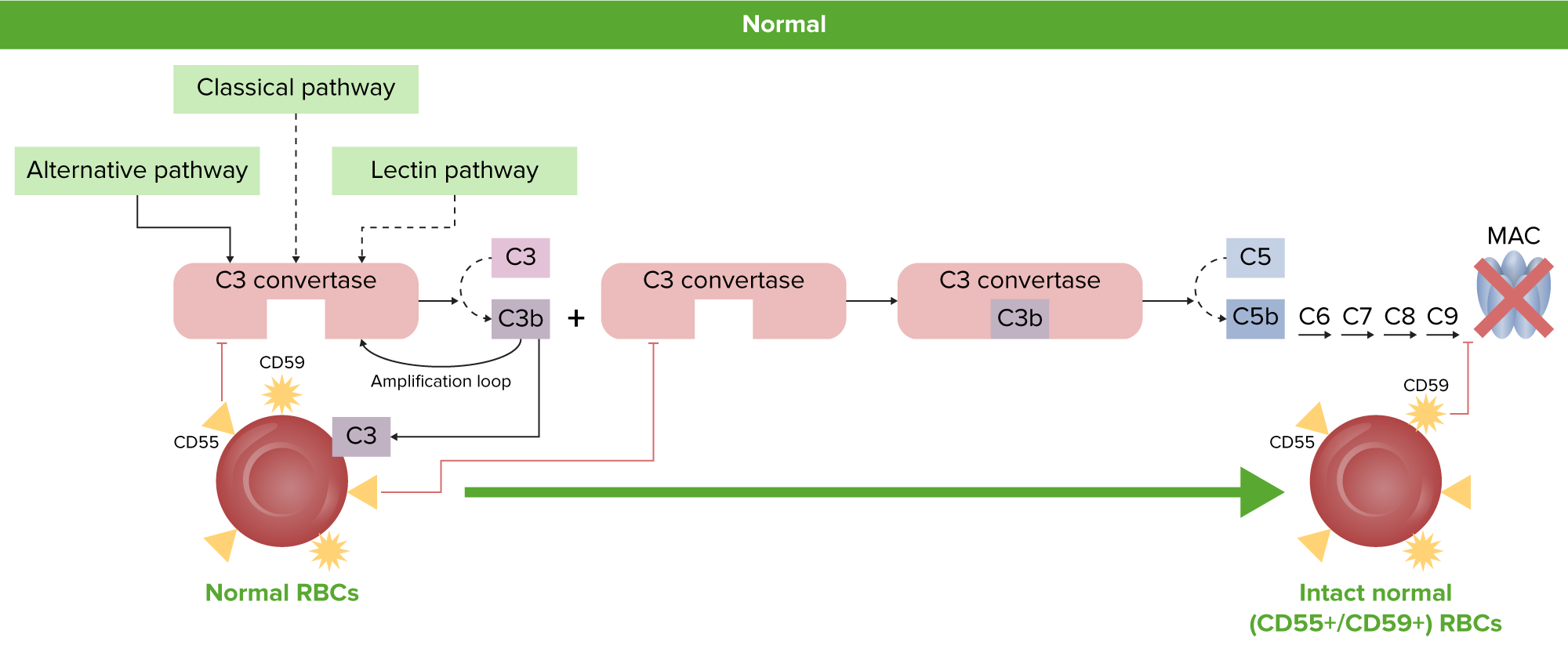
Fisiología normal de los eritrocitos:
Los eritrocitos normales están protegidos de la activación del complemento y de la hemólisis por las proteínas reguladoras del complemento ancladas a GPI, CD55 y CD59.
GPI: glucosilfosfatidilinositol
MAC: complejo de ataque a la membrana
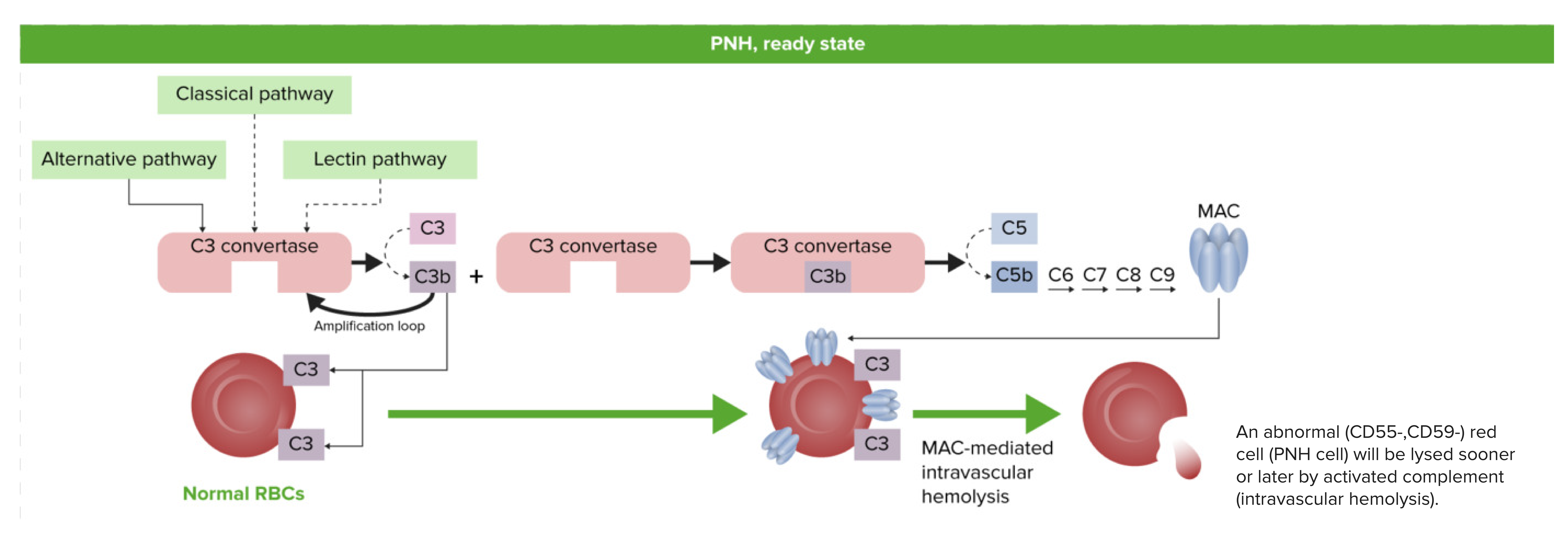
Fisiología de la hemoglobinuria paroxística nocturna (HPN) en estado estacionario:
Incluso en ausencia de inflamación, infección u otro desencadenante, los eritrocitos con GPI reducido o inexistente en su superficie, son susceptibles de ser atacados por el complemento mediante la activación espontánea del mismo a partir de una cantidad trivial, pero constantemente presente de C3 activado (el “ralentí de C3”). La hemólisis intravascular se produce debido a la formación de un complejo de ataque a la membrana (MAC, por sus siglas en inglés), que provoca una alteración focal y agujeros en la membrana. Si se activa complemento adicional a través de la vía clásica antígeno-anticuerpo, (por ejemplo, por una infección), la hemólisis aumentará; esto corresponde a exacerbación paroxística de la enfermedad.
GPI: glucosilfosfatidilinositol

Muestra de orina de un paciente con HPN (izquierda) y de un control sano (derecha).
La orina del paciente es roja debido a la hemólisis intravascular con hemoglobinuria. La hemólisis se produce durante todo el día, no solo por la noche.
Los LOS Neisseria neutrófilos también presentan una deficiencia de proteínas ancladas a glucosilfosfatidilinositol:
Los LOS Neisseria factores mediados por plaquetas incluyen:
La trombosis en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum sitios inusuales (venas hepáticas, mesentéricas, cerebrales, cutáneas) es típica de la HPN.
La insuficiencia renal es progresiva y multifactorial.
Las exacerbaciones de la hemólisis pueden deberse a:
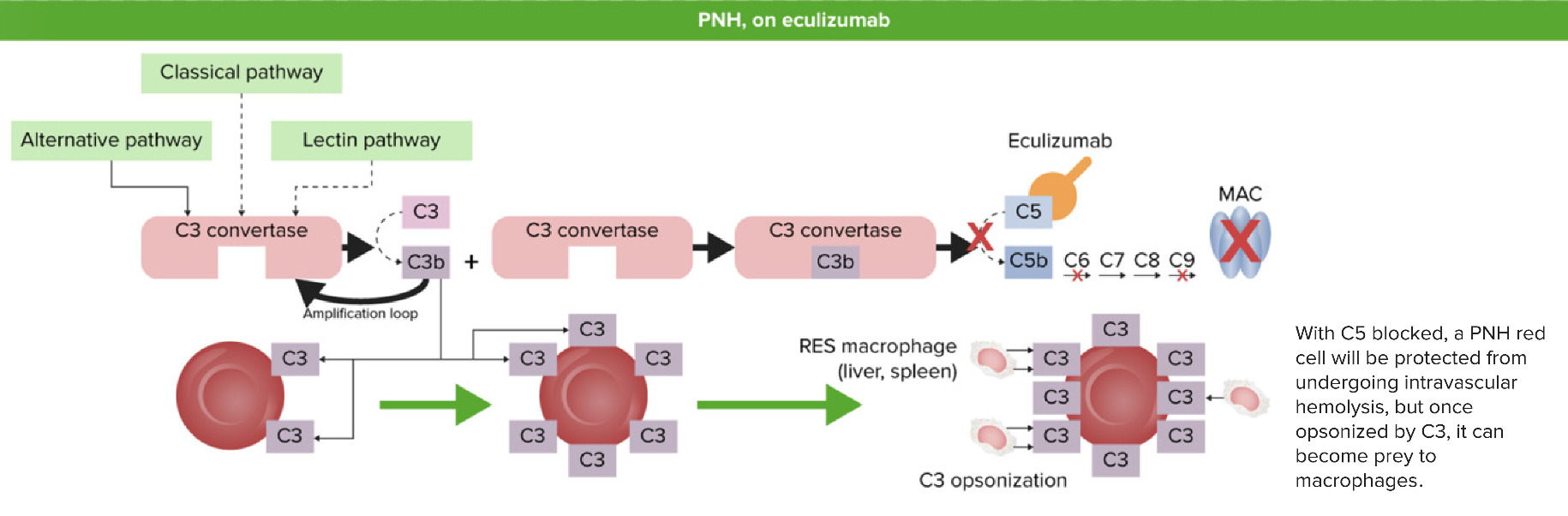
Hemoglobinuria paroxística nocturna tras recibir eculizumab:
Cuando C5 está bloqueado (inhibido) por el eculizumab, la convertasa C5 no puede formar C5b a partir de la escisión de C5. No se forma el complejo de ataque a la membrana (MAC) y no se produce hemólisis intravascular. Sin embargo, los fragmentos de C3 liberados anteriormente en el proceso de activación recubrirán los eritrocitos desprotegidos y provocarán hemólisis extravascular por parte del sistema reticuloendotelial en el hígado y el bazo.
Estos síntomas se deben a la disminución del NO circulante: