Hypocoagulable conditions, also known as bleeding disorders or bleeding diatheses, are a diverse group of diseases that result in abnormal hemostasis Hemostasis Hemostasis refers to the innate, stepwise body processes that occur following vessel injury, resulting in clot formation and cessation of bleeding. Hemostasis occurs in 2 phases, namely, primary and secondary. Primary hemostasis involves forming a plug that stops the bleeding temporarily. Secondary hemostasis involves the activation of the coagulation cascade. Hemostasis. Hemostasis Hemostasis Hemostasis refers to the innate, stepwise body processes that occur following vessel injury, resulting in clot formation and cessation of bleeding. Hemostasis occurs in 2 phases, namely, primary and secondary. Primary hemostasis involves forming a plug that stops the bleeding temporarily. Secondary hemostasis involves the activation of the coagulation cascade. Hemostasis is the innate, stepwise process resulting in bleeding cessation from a damaged blood vessel. Physiologic hemostasis Hemostasis Hemostasis refers to the innate, stepwise body processes that occur following vessel injury, resulting in clot formation and cessation of bleeding. Hemostasis occurs in 2 phases, namely, primary and secondary. Primary hemostasis involves forming a plug that stops the bleeding temporarily. Secondary hemostasis involves the activation of the coagulation cascade. Hemostasis is dependent on the integrity of endothelial cells and subendothelial Subendothelial Membranoproliferative Glomerulonephritis matrix, platelets Platelets Platelets are small cell fragments involved in hemostasis. Thrombopoiesis takes place primarily in the bone marrow through a series of cell differentiation and is influenced by several cytokines. Platelets are formed after fragmentation of the megakaryocyte cytoplasm. Platelets: Histology, and coagulation factors Coagulation factors Endogenous substances, usually proteins, that are involved in the blood coagulation process. Hemostasis. The hypocoagulable states result from abnormalities in one or more of these contributors, resulting in ineffective thrombosis Thrombosis Formation and development of a thrombus or blood clot in the blood vessel. Epidemic Typhus and bleeding.
Last updated: Dec 15, 2025
Hypocoagulable conditions, also known as bleeding disorders or bleeding diatheses, are a diverse group of diseases that result in abnormal hemostasis Hemostasis Hemostasis refers to the innate, stepwise body processes that occur following vessel injury, resulting in clot formation and cessation of bleeding. Hemostasis occurs in 2 phases, namely, primary and secondary. Primary hemostasis involves forming a plug that stops the bleeding temporarily. Secondary hemostasis involves the activation of the coagulation cascade. Hemostasis and increased bleeding risk.
Physiologic hemostasis Hemostasis Hemostasis refers to the innate, stepwise body processes that occur following vessel injury, resulting in clot formation and cessation of bleeding. Hemostasis occurs in 2 phases, namely, primary and secondary. Primary hemostasis involves forming a plug that stops the bleeding temporarily. Secondary hemostasis involves the activation of the coagulation cascade. Hemostasis is dependent on normal structure and function of:
The following is a summary of the process:
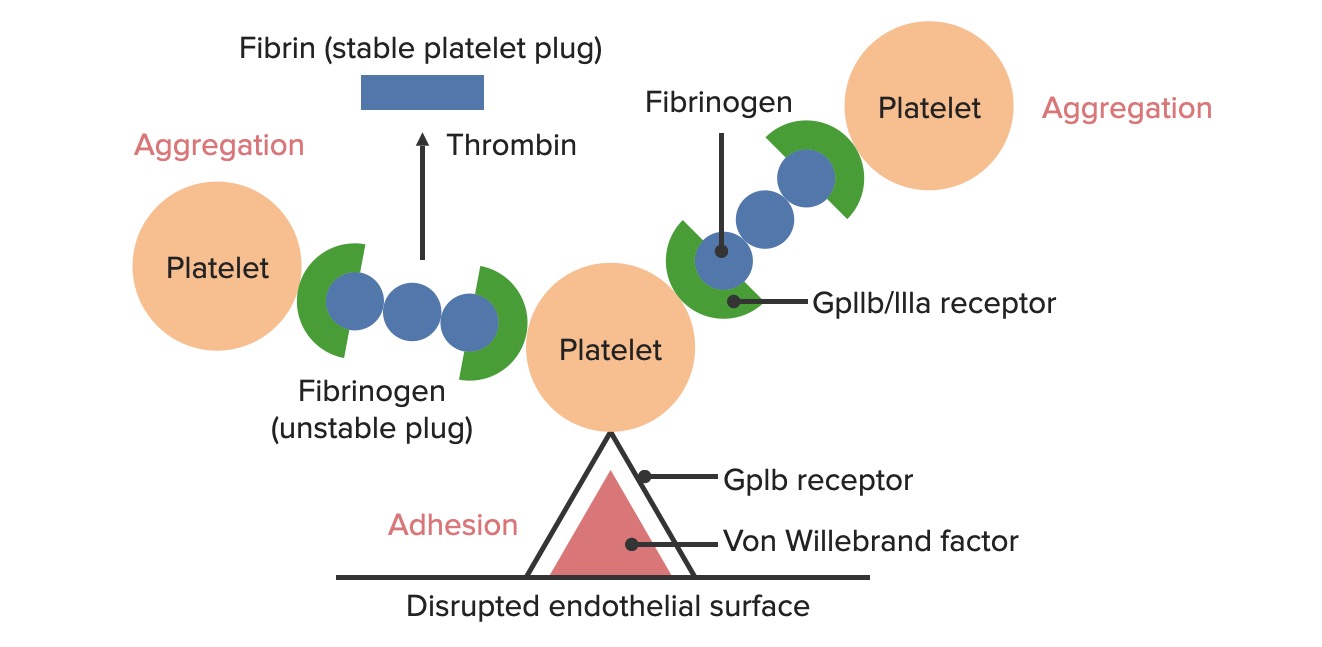
Formation of the temporary hemostatic plug:
The disrupted endothelial surface exposes von Willebrand Factor (vWF) to the passing blood. Platelets bind to the vWF via their GpIb receptors and are activated. Platelet activation triggers them to secrete adenosine diphosphate (ADP), which stimulates the expression of the GpIIb/IIIa receptors on the platelets. The GpIIb/IIIa receptors bind to fibrinogen, which is able to bind a platelet on each end, causing platelets to aggregate. As more platelets are bound to one another, the platelet plug is generated. As the coagulation cascade is activated, thrombin converts the weaker fibrinogen into the stronger fibrin, creating a much more stable clot.
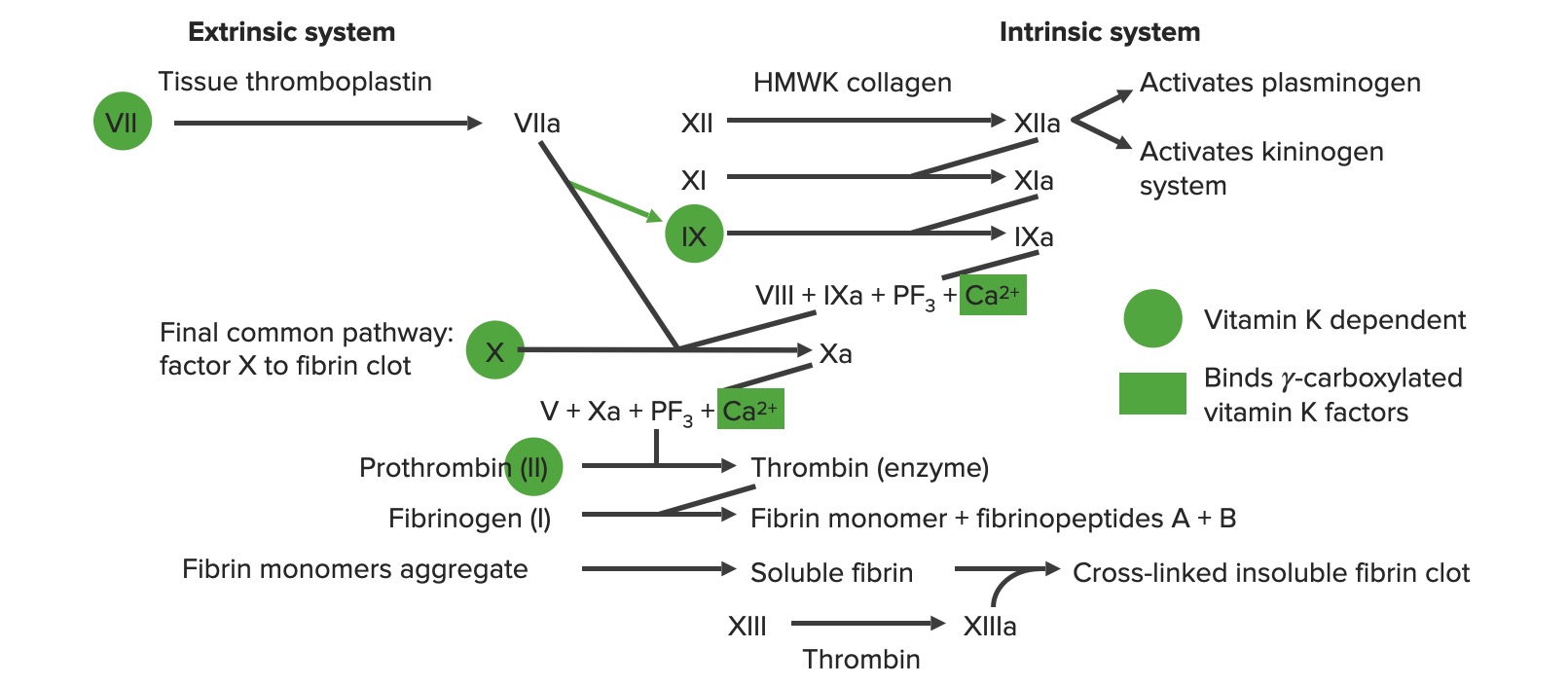
Overview of the coagulation cascade
a: activated form
PF3: platelet factor 3 (phospholipids)
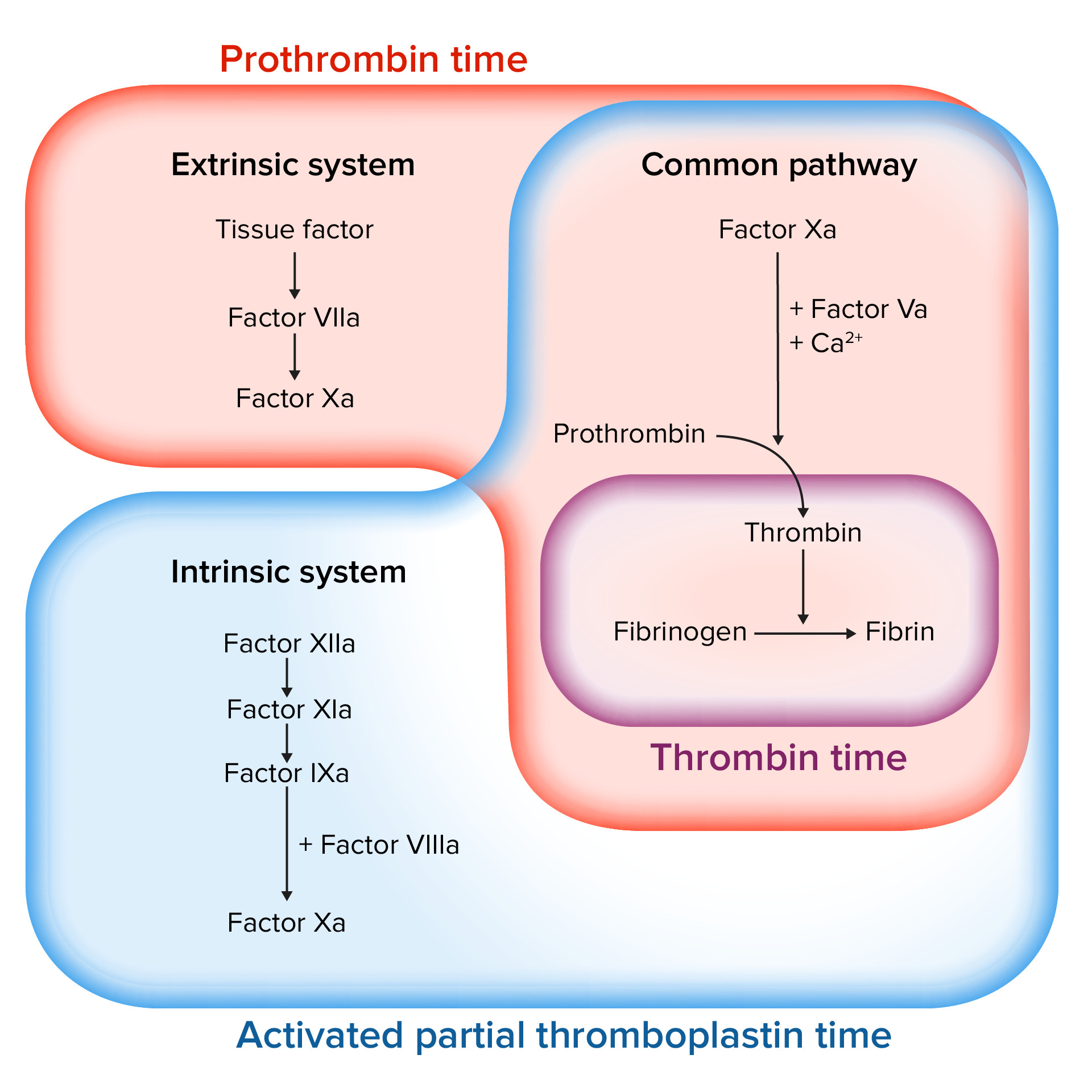
Assessing the coagulation cascade
Image by Lecturio.The following conditions can lead to a hypocoagulable state.
| Inherited conditions | Acquired conditions | |
|---|---|---|
| Vessel wall disorders |
|
|
| Platelet disorders |
|
|
| Coagulation disorders | Hemophilias A, B, and C |
|
| Mixed disorders | Von Willebrand disease Von Willebrand disease Von Willebrand disease (vWD) is a bleeding disorder characterized by a qualitative or quantitative deficiency of von Willebrand factor (vWF). Von Willebrand factor is a multimeric protein involved in the plate adhesion phase of hemostasis by forming a bridge between platelets and damaged portions of the vessel wall. Von Willebrand Disease | Disseminated intravascular coagulation Disseminated intravascular coagulation Disseminated intravascular coagulation (DIC) is a condition characterized by systemic bodywide activation of the coagulation cascade. This cascade results in both widespread microvascular thrombi contributing to multiple organ dysfunction and consumption of clotting factors and platelets, leading to hemorrhage. Disseminated Intravascular Coagulation |
| Medications |
|
|
A hypocoagulable state may present in the following ways:
| Cutaneous bleeding | Mucosal bleeding Mucosal bleeding Chédiak-Higashi Syndrome | Internal bleeding | Onset of bleeding after trauma | |
|---|---|---|---|---|
| Platelet disorders | ✓ | ✓ | – | Early (immediately) |
| Vessel disorders | ✓ | – | – | – |
| Disorders of the coagulation cascade Coagulation cascade The coagulation cascade is a series of reactions that ultimately generates a strong, cross-linked fibrin clot. Hemostasis | – | – | ✓ | Late (minutes to hours) |
Bleeding disorders may stem from abnormalities of the vessel wall.
Inherited disorders of the connective tissue Connective tissue Connective tissues originate from embryonic mesenchyme and are present throughout the body except inside the brain and spinal cord. The main function of connective tissues is to provide structural support to organs. Connective tissues consist of cells and an extracellular matrix. Connective Tissue: Histology matrix can cause vascular fragility, leading to frequent vessel injury.
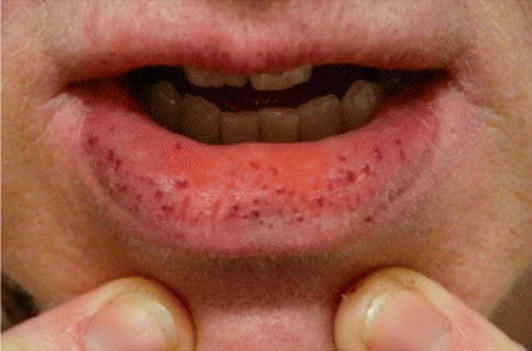
Hereditary hemorrhagic telangiectasia
Image: “Clinical manifestations of HHT” by Latino et al. License: CC BY 4.0, cropped by Lecturio.The following acquired conditions may lead to vascular fragility and increase vessel injury:
Inherited disorders:
Acquired conditions: