


Os distúrbios de hipocoagulabilidade, também conhecidas como distúrbios hemorrágicos ou diáteses hemorrágicas, são um grupo diverso de doenças que resultam em hemostase anormal. A hemostase é o processo inato e gradual que resulta na cessação da hemorragia de um vaso sanguíneo danificado. A hemostase fisiológica depende da integridade das células endoteliais e da matriz subendotelial, das plaquetas e dos fatores de coagulação. Os estados de hipocoagulabilidade resultam de anomalias em um ou mais MAIS Androgen Insensitivity Syndrome destes fatores contributivos, resultando em trombose ineficaz e hemorragia.
Last updated: Dec 15, 2025
Os distúrbios de hipocoagulabilidade, também conhecidos como distúrbios hemorrágicos ou diáteses hemorrágicas, são um grupo diverso de doenças que resultam em hemostase anormal e aumento do risco de hemorragia.
A hemostase fisiológica depende da estrutura e função normais de:
Abaixo encontra-se um resumo do processo:
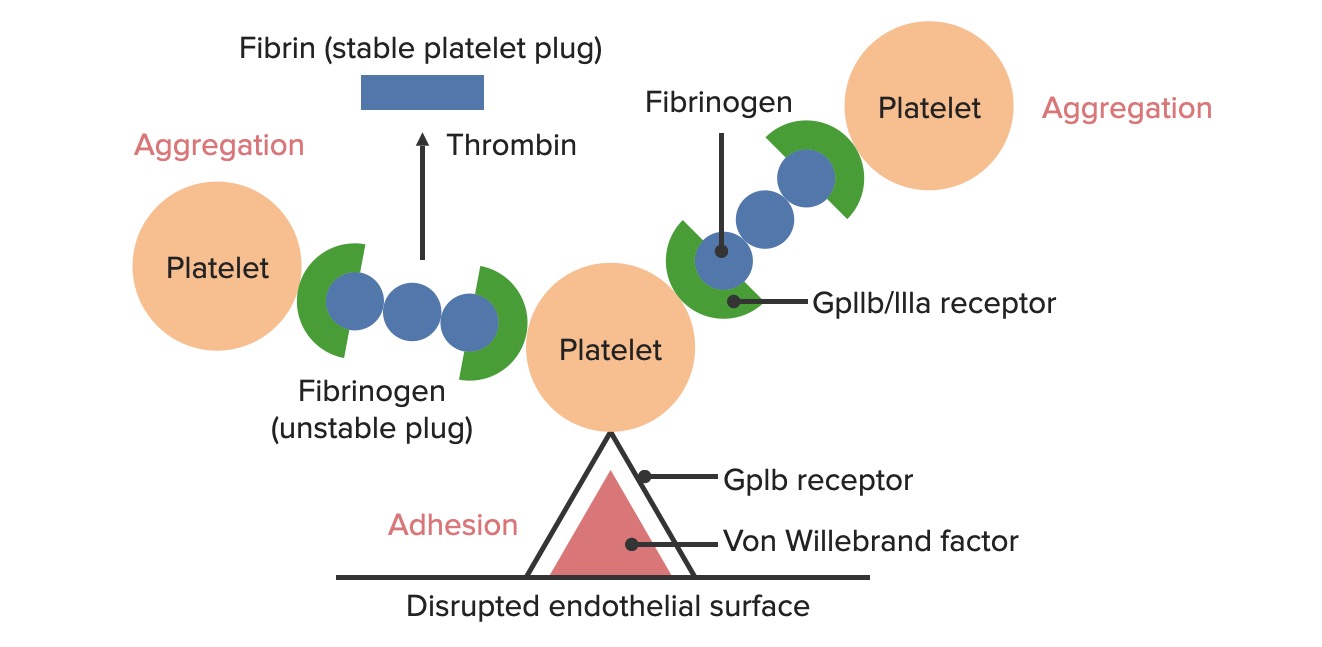
Formação do tampão hemostático temporário:
A superfície endotelial danificada expõe o fator de von Willebrand (FvW) ao sangue que passa. As plaquetas ligam-se ao FvW através dos seus recetores GpIb e são ativadas. A ativação plaquetária desencadeia a secreção de difosfato de adenosina (ADP), que estimula a expressão dos recetores GpIIb/IIIa nas plaquetas. Os recetores GpIIb/IIIa ligam-se ao fibrinogénio, que é capaz de ligar uma plaqueta em cada extremidade, fazendo com que as plaquetas se agreguem. À medida que mais plaquetas são ligadas umas às outras, gera-se o tampão plaquetário. À medida que a cascata de coagulação é ativada, a trombina converte o fibrinogénio mais frágil em fibrina mais forte, criando um coágulo muito mais estável.
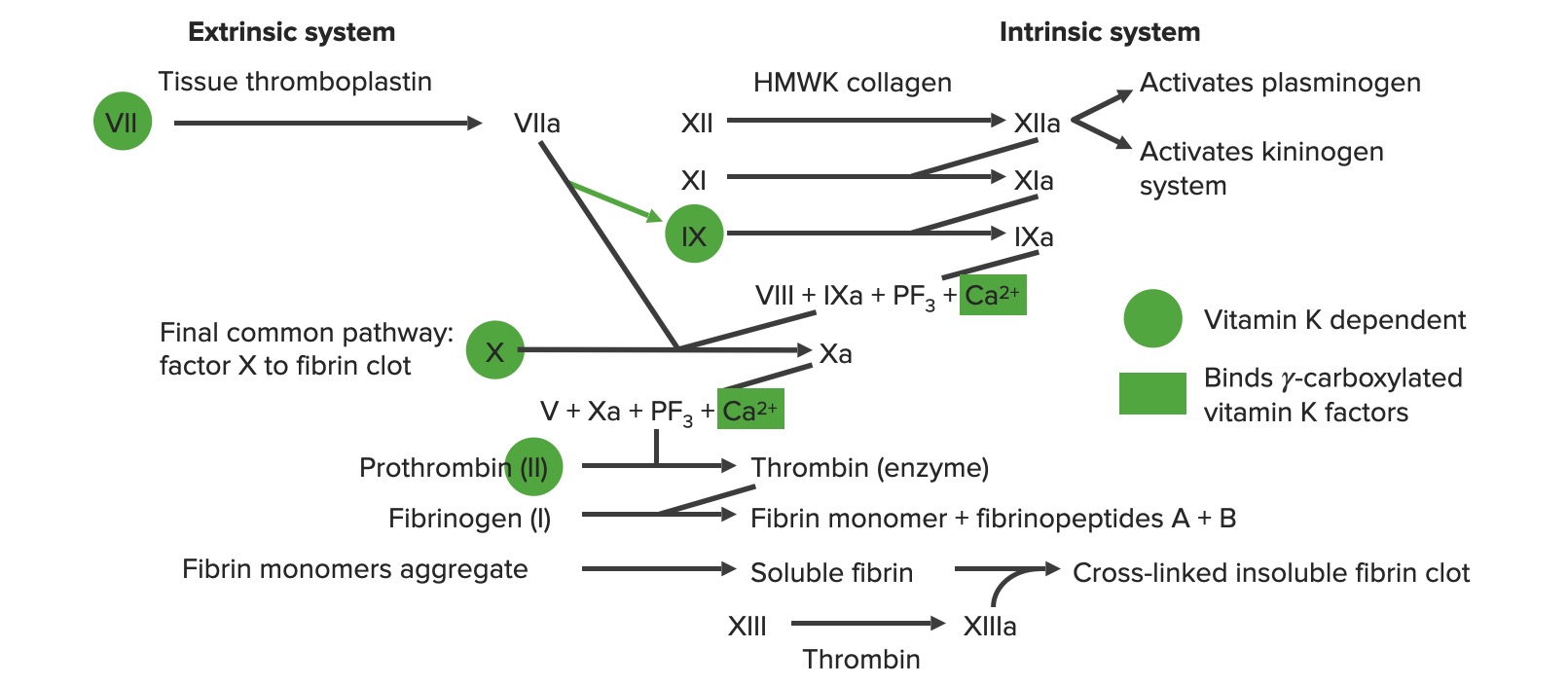
Visão geral da cascata de coagulação
a: formulário ativado
PF3 : fator plaquetário 3 (fosfolipídios)
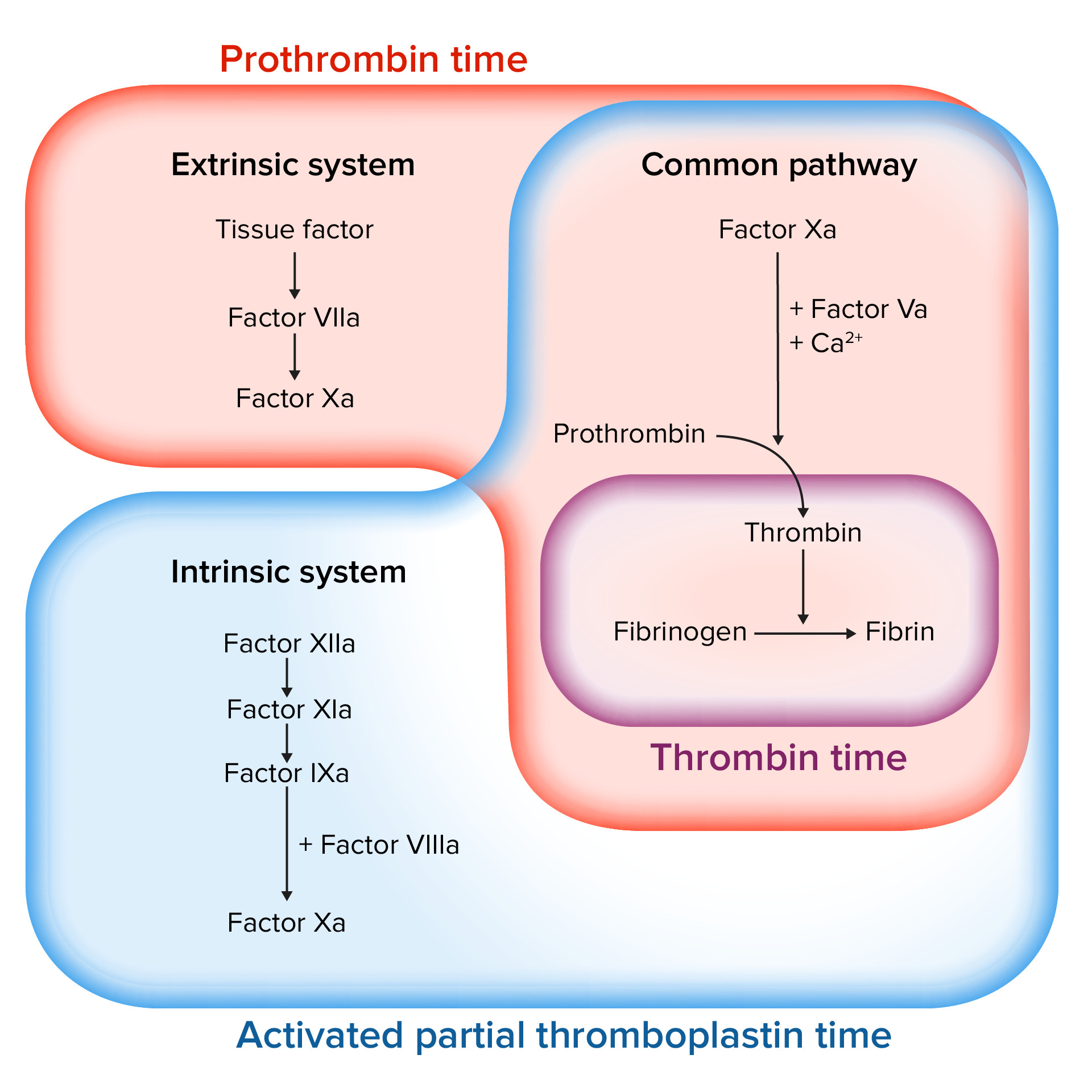
Avaliação da cascata de coagulação
Imagem por Lecturio.As seguintes condições podem levar a um estado de hipocoagulabilidade.
| Doenças herdadas | Doenças adquiridas | |
|---|---|---|
| Distúrbios da parede do vaso |
|
|
| Distúrbios das plaquetas |
|
|
| Distúrbios da coagulação | Hemofilias A, B e C |
|
| Distúrbios mistos | Doença de von Willebrand | Coagulação intravascular disseminada |
| Fármacos |
|
|
Um estado de hipocoagulabilidade pode se apresentar das seguintes formas:
| Hemorragia cutânea | Hemorragia das mucosas | Hemorragia interna | Início de hemorragia após o trauma | |
|---|---|---|---|---|
| Distúrbios das plaquetas | ✓ | ✓ | – | Precoce (imediatamente) |
| Distúrbios dos vasos | ✓ | – | – | – |
| Distúrbios da cascata de coagulação | – | – | ✓ | Tardia (minutos a horas) |
Os distúrbios hemorrágicos podem resultar de alterações das paredes dos vasos.
Os distúrbios hereditários da matriz do tecido conjuntivo podem causar fragilidade vascular, levando a lesões vasculares frequentes.
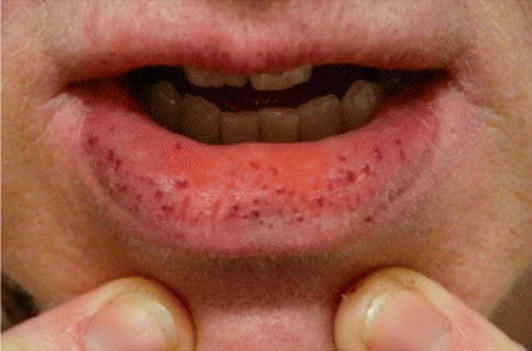
Telangiectasia hemorrágica hereditária
Imagem: “Clinical manifestations of HHT” por Latino et al. Licença: CC BY 4.0, cortado por Lecturio.As seguintes condições adquiridas podem levar à fragilidade vascular e aumentar as lesões vasculares:
Distúrbios hereditários:
Doenças adquiridas:
