Las afecciones hipocoagulables, también conocidas como trastornos hemorrágicos o diátesis hemorrágicas, son un grupo diverso de enfermedades que dan lugar a una hemostasia anormal. La hemostasia es el proceso innato y escalonado que da lugar al AL Amyloidosis cese de la hemorragia de un vaso sanguíneo dañado. La hemostasia fisiológica depende de la integridad de las células endoteliales y la matriz subendotelial, las plaquetas y los LOS Neisseria factores de coagulación. Los LOS Neisseria estados hipocoagulables son el resultado de anomalías en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum uno o varios de estos factores contribuyentes, lo que da lugar a trombosis inefectivas y hemorragias.
Last updated: Dec 15, 2025
Las afecciones hipocoagulables, también conocidas como trastornos hemorrágicos o diátesis hemorrágicas, son un grupo diverso de enfermedades que provocan una hemostasia anormal y un mayor riesgo de hemorragia.
La hemostasia fisiológica depende una normal estructura y funcionamiento de:
A continuación se presenta un resumen del proceso:
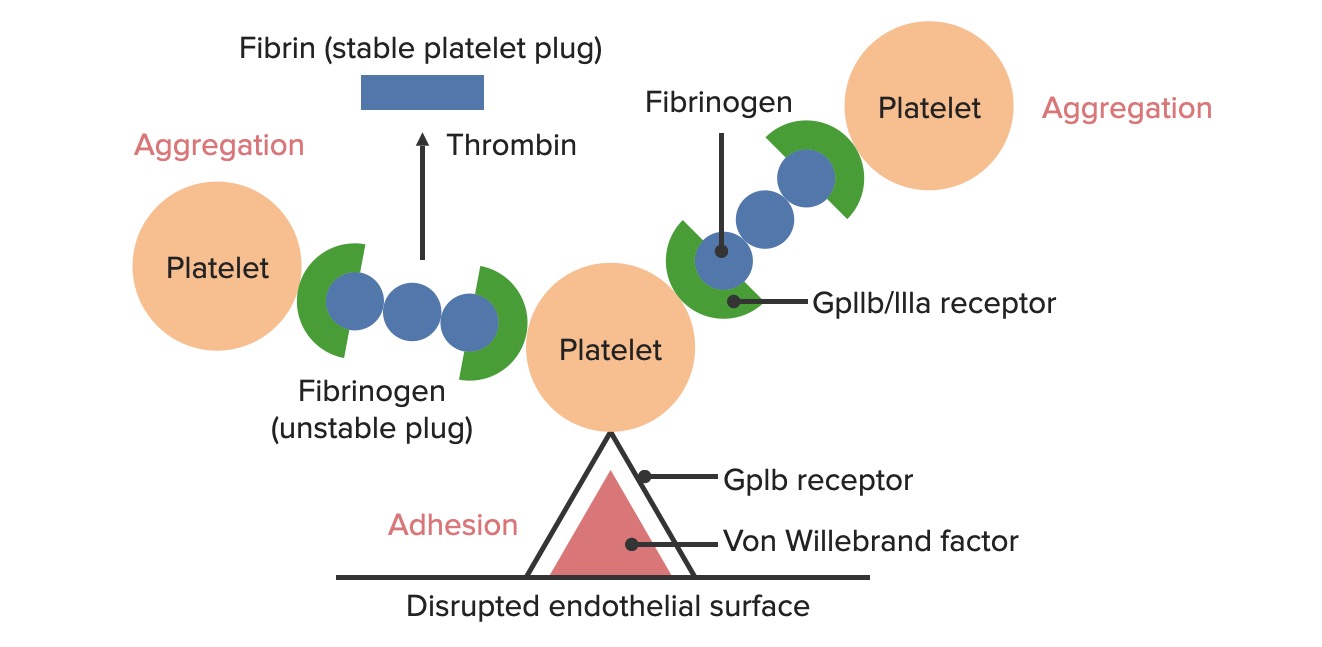
Formación del tapón hemostático temporal:
La superficie endotelial alterada expone el FVW al paso de la sangre. Las plaquetas se unen al FVW a través de sus receptores GpIb y se activan. La activación de las plaquetas las lleva a secretar adenosín difosfato (ADP), lo que estimula la expresión de los receptores GpIIb/IIIa en las plaquetas. Los receptores GpIIb/IIIa se unen al fibrinógeno, que es capaz de unir una plaqueta en cada extremo, provocando la agregación de las plaquetas. A medida que se unen más plaquetas entre sí, se genera el tapón plaquetario. Al activarse la cascada de la coagulación, la trombina convierte el fibrinógeno débil en fibrina más fuerte, creando un coágulo mucho más estable.
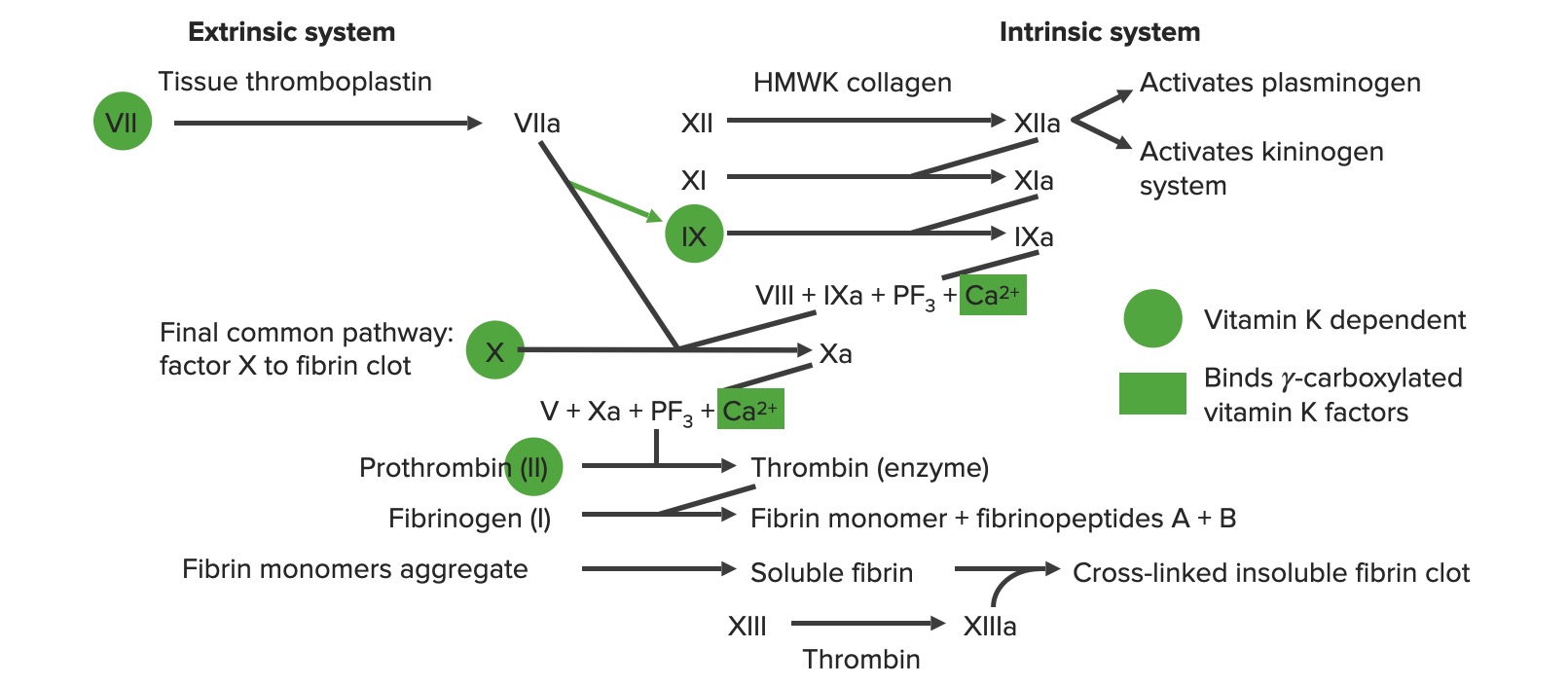
Visión general de la cascada de coagulación
a: forma activada
PF3: factor plaquetario 3 (fosfolípidos)
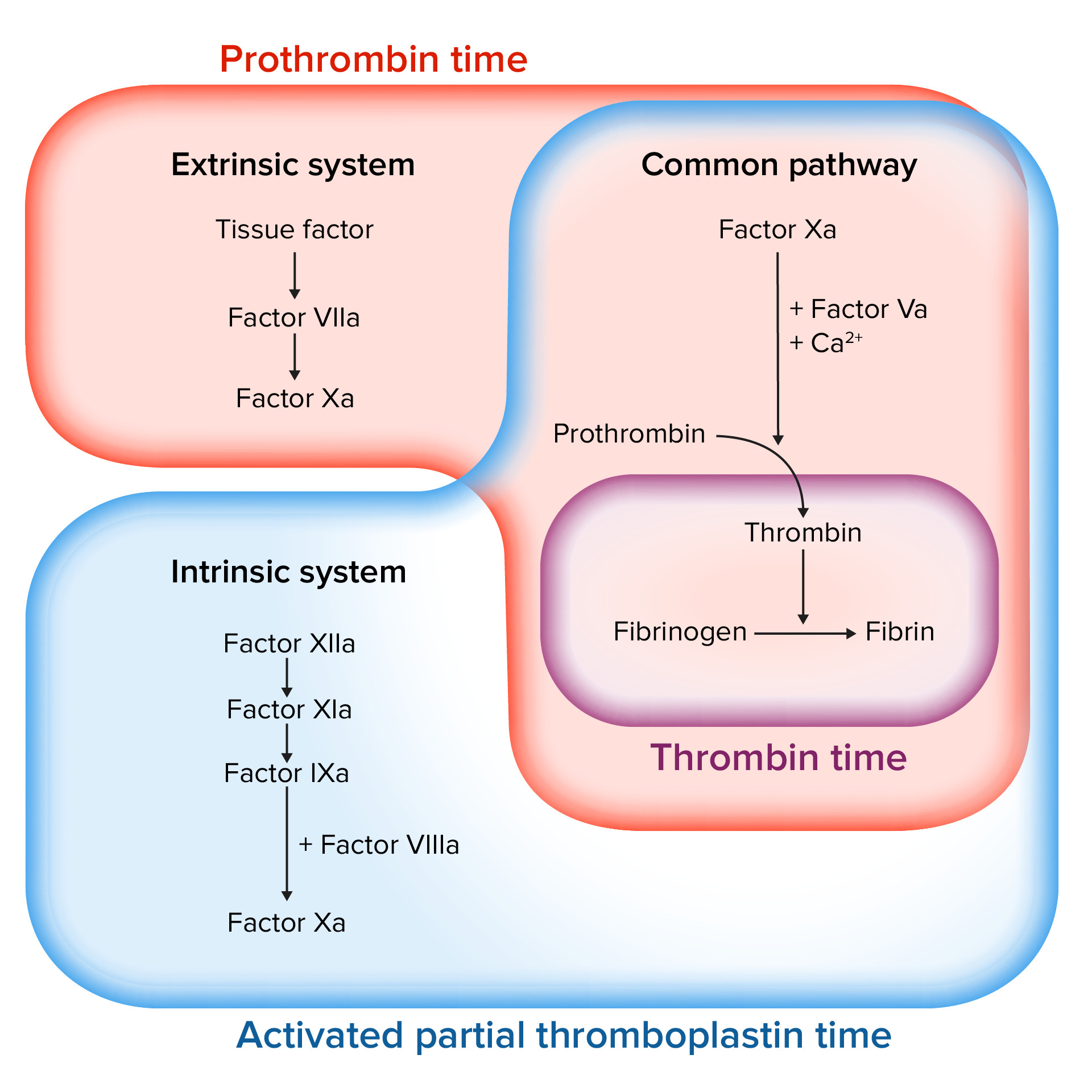
Evaluación de la cascada de coagulación
Imagen por Lecturio.Las siguientes afecciones pueden conducir a un estado hipocoagulable.
| Afecciones heredadas | Afecciones adquiridas | |
|---|---|---|
| Trastornos de la pared del vaso |
|
|
| Trastornos plaquetarios |
|
|
| Trastornos de la coagulación | Hemofilias A, B y C |
|
| Trastornos mixtos | Enfermedad de Von Willebrand | Coagulación intravascular diseminada |
| Medicamentos |
|
|
Un estado hipocoagulable puede presentarse de las siguientes maneras:
| Hemorragia cutánea | Hemorragia de la mucosa | Hemorragia interna | Inicio de hemorragia después de un traumatismo | |
|---|---|---|---|---|
| Trastornos plaquetarios | ✓ | ✓ | – | Temprano (inmediatamente) |
| Trastornos vasculares | ✓ | – | – | – |
| Trastornos de la cascada de la coagulación | – | – | ✓ | Tarde (de minutos a horas) |
Los LOS Neisseria trastornos hemorrágicos pueden tener su origen en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum anomalías de la pared de los LOS Neisseria vasos.
Los LOS Neisseria trastornos hereditarios de la matriz del tejido conectivo pueden causar fragilidad vascular, lo que provoca frecuentes lesiones en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum los LOS Neisseria vasos.
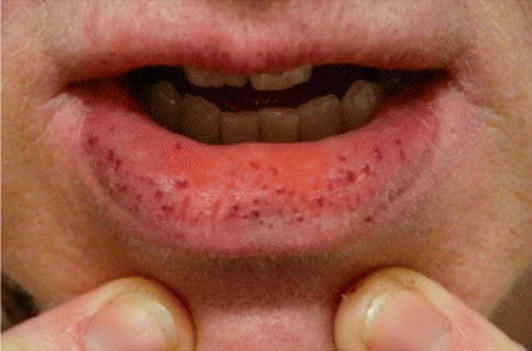
Telangiectasia hemorrágica hereditaria
Imagen: “Clinical manifestations of HHT” por Latino et al. Licencia: CC BY 4.0, recortada por Lecturio.Las siguientes condiciones adquiridas pueden conducir a la fragilidad vascular y aumentar la lesión de los LOS Neisseria vasos:
Trastornos hereditarios:
Condiciones adquiridas: