Ehlers-Danlos syndrome (EDS) is a heterogeneous group of inherited connective tissue Connective tissue Connective tissues originate from embryonic mesenchyme and are present throughout the body except inside the brain and spinal cord. The main function of connective tissues is to provide structural support to organs. Connective tissues consist of cells and an extracellular matrix. Connective Tissue: Histology disorders that are characterized by hyperextensible skin Skin The skin, also referred to as the integumentary system, is the largest organ of the body. The skin is primarily composed of the epidermis (outer layer) and dermis (deep layer). The epidermis is primarily composed of keratinocytes that undergo rapid turnover, while the dermis contains dense layers of connective tissue. Skin: Structure and Functions, hypermobile joints, and fragility of the skin Skin The skin, also referred to as the integumentary system, is the largest organ of the body. The skin is primarily composed of the epidermis (outer layer) and dermis (deep layer). The epidermis is primarily composed of keratinocytes that undergo rapid turnover, while the dermis contains dense layers of connective tissue. Skin: Structure and Functions and connective tissue Connective tissue Connective tissues originate from embryonic mesenchyme and are present throughout the body except inside the brain and spinal cord. The main function of connective tissues is to provide structural support to organs. Connective tissues consist of cells and an extracellular matrix. Connective Tissue: Histology. The syndrome is due to genetic defects that affect collagen Collagen A polypeptide substance comprising about one third of the total protein in mammalian organisms. It is the main constituent of skin; connective tissue; and the organic substance of bones (bone and bones) and teeth (tooth). Connective Tissue: Histology processing and synthesis Synthesis Polymerase Chain Reaction (PCR). There are many subtypes that vary in terms of inheritance, severity, and clinical presentation. The diagnosis is mainly clinical and is supported by molecular testing for many subtypes. There is no curative treatment. Management involves understanding the many complications of the disease and the interdisciplinary prevention and treatment of specific symptoms.
Last updated: Dec 15, 2025
Ehlers-Danlos syndrome (EDS) is a group of 13 inherited connective tissue Connective tissue Connective tissues originate from embryonic mesenchyme and are present throughout the body except inside the brain and spinal cord. The main function of connective tissues is to provide structural support to organs. Connective tissues consist of cells and an extracellular matrix. Connective Tissue: Histology disorders affecting collagen Collagen A polypeptide substance comprising about one third of the total protein in mammalian organisms. It is the main constituent of skin; connective tissue; and the organic substance of bones (bone and bones) and teeth (tooth). Connective Tissue: Histology and the extracellular matrix Extracellular matrix A meshwork-like substance found within the extracellular space and in association with the basement membrane of the cell surface. It promotes cellular proliferation and provides a supporting structure to which cells or cell lysates in culture dishes adhere. Hypertrophic and Keloid Scars (structure, processing, or related proteins Proteins Linear polypeptides that are synthesized on ribosomes and may be further modified, crosslinked, cleaved, or assembled into complex proteins with several subunits. The specific sequence of amino acids determines the shape the polypeptide will take, during protein folding, and the function of the protein. Energy Homeostasis). The syndrome is clinically heterogeneous and can affect the skin Skin The skin, also referred to as the integumentary system, is the largest organ of the body. The skin is primarily composed of the epidermis (outer layer) and dermis (deep layer). The epidermis is primarily composed of keratinocytes that undergo rapid turnover, while the dermis contains dense layers of connective tissue. Skin: Structure and Functions, joints, blood vessels, and other tissues and is characterized by the following:
This list of classifications is not exhaustive; it includes the most common, clinically significant subtypes.
Skin Skin The skin, also referred to as the integumentary system, is the largest organ of the body. The skin is primarily composed of the epidermis (outer layer) and dermis (deep layer). The epidermis is primarily composed of keratinocytes that undergo rapid turnover, while the dermis contains dense layers of connective tissue. Skin: Structure and Functions:
Musculoskeletal:
Cardiovascular:
Pulmonary:
GI:
Ophthalmic:
Obstetric/gynecologic:
Miscellaneous:
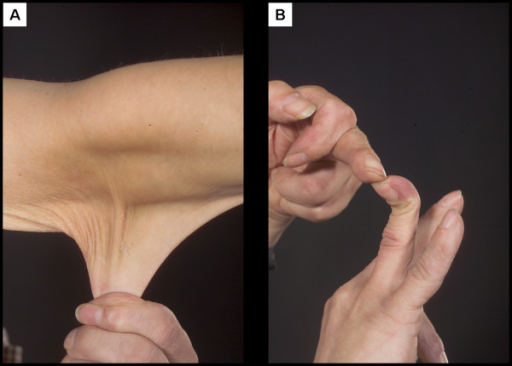
Examples of a patient with Ehlers-Danlos syndrome
A: Prominent hyperextensible skin
B: Hypermobile joints
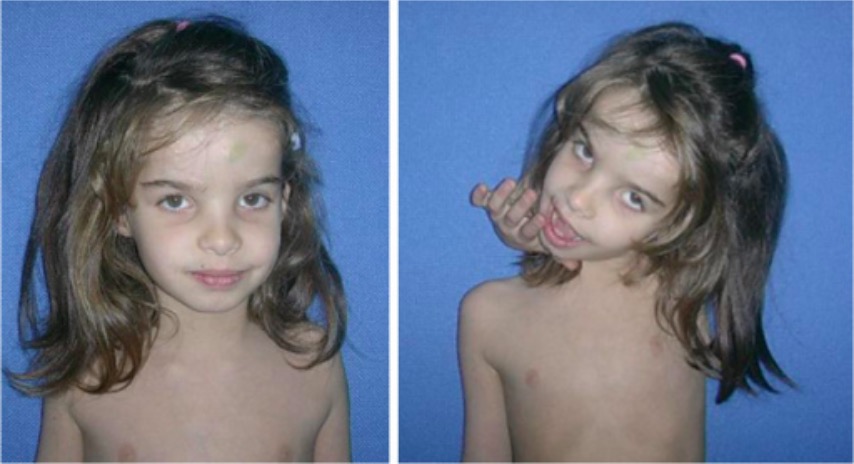
Young girl with Ehlers-Danlos syndrome:
Joint laxity of the cervical spine is notable.
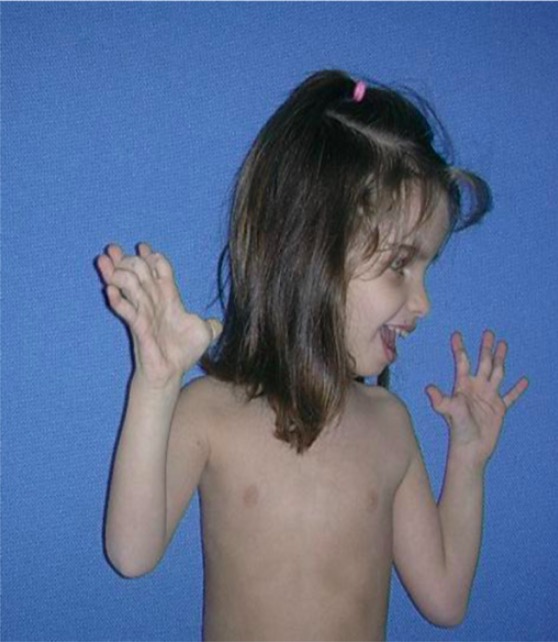
Young girl with Ehlers-Danlos syndrome:
Joint laxity of the fingers is notable.
Ehlers-Danlos syndrome is generally diagnosed on the basis of patient history and clinical findings.