


A amiloidose é uma doença causada pela deposição anómala de fibrilhas, compostas por várias subunidades de proteína de baixo peso molecular com enovelamento deficiente, no tecido extracelular. Estas proteínas são frequentemente subprodutos de outros processos patológicos (por exemplo, mieloma múltiplo). Estas proteínas com enovelamento deficiente podem depositar-se em diferentes tecidos, interferir nas funções normais dos órgãos e causar doenças específicas dos tecidos (por exemplo, a amiloidose renal causa proteinúria). O diagnóstico é estabelecido clinicamente e confirmado com a biópsia do tecido. O tratamento deve ser orientado para a causa subjacente e para a redução do depósito de amiloide.
Last updated: Dec 15, 2025
A amiloidose é uma doença causada pela deposição anómala de fibrilhas, compostas por várias subunidades de proteína de baixo peso molecular com enovelamento deficiente, no tecido extracelular.
Amiloidose sistémica:
Amiloidose localizada:
Amiloidose hereditária:
| Doença | Proteína amiloide | Órgãos envolvidos | Especificidades |
|---|---|---|---|
| Amiloidose primária | AL AL Amyloidosis | Envolvimento sistémico:
|
|
| Amiloidose secundária | AA AA Amyloidosis | ||
| Amiloidose associada à hemodiálise | Aβ₂M |
|
|
| Doença | Proteína amiloide | Órgãos envolvidos | Especificidades |
|---|---|---|---|
| Amiloidose cerebral | Amiloide Aβ |
|
|
| Carcinoma medular da tiroide | ACal ACal Amyloidosis | Glândula tiroide | Clinicamente insignificante |
| Diabetes Diabetes Diabetes mellitus (DM) is a metabolic disease characterized by hyperglycemia and dysfunction of the regulation of glucose metabolism by insulin. Type 1 DM is diagnosed mostly in children and young adults as the result of autoimmune destruction of β cells in the pancreas and the resulting lack of insulin. Type 2 DM has a significant association with obesity and is characterized by insulin resistance. Diabetes Mellitus mellitus tipo 2 | AIAPP | Ilhéus pancreáticos de Langerhans | Associada a insulinoma Insulinoma A benign tumor of the pancreatic beta cells. Insulinoma secretes excess insulin resulting in hypoglycemia. Pancreatic Neuroendocrine Tumors (PanNETs) |
| Amiloidose auricular isolada | AANP AANP Amyloidosis | Aurículas | Associada ao envelhecimento |
| Doença | Proteína amiloide | Órgãos envolvidos | Especificidades |
|---|---|---|---|
| Febre mediterrânica familiar | AA AA Amyloidosis |
|
|
| Neuropatia amiloide familiar | ATTR | Nervos periféricos |
|
| Cardiomiopatia amiloide familiar | ATTR | Ventrículos |
|
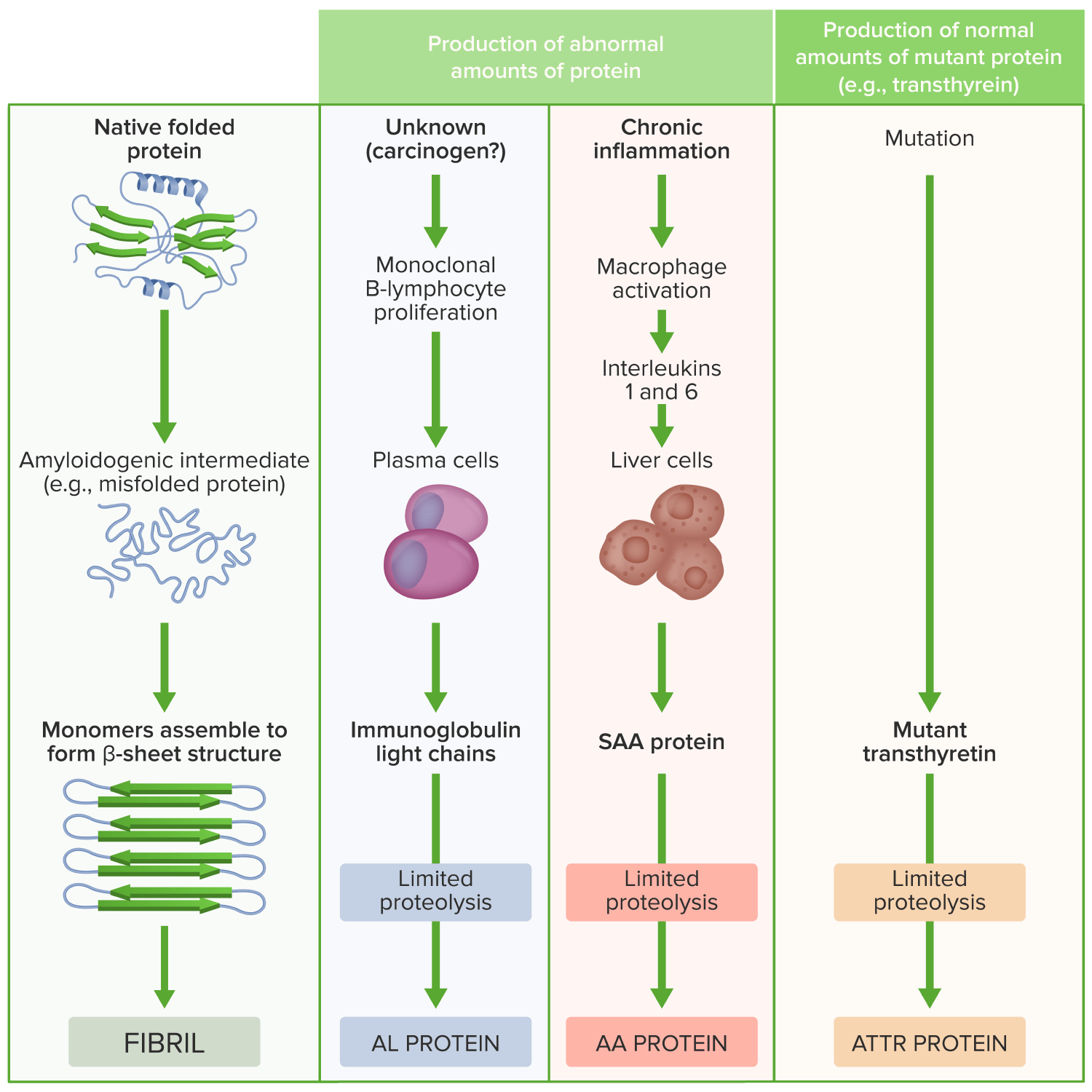
Patogénese da amiloidose
Imagem por Lecturio.A apresentação clínica depende do órgão/orgãos envolvidos.
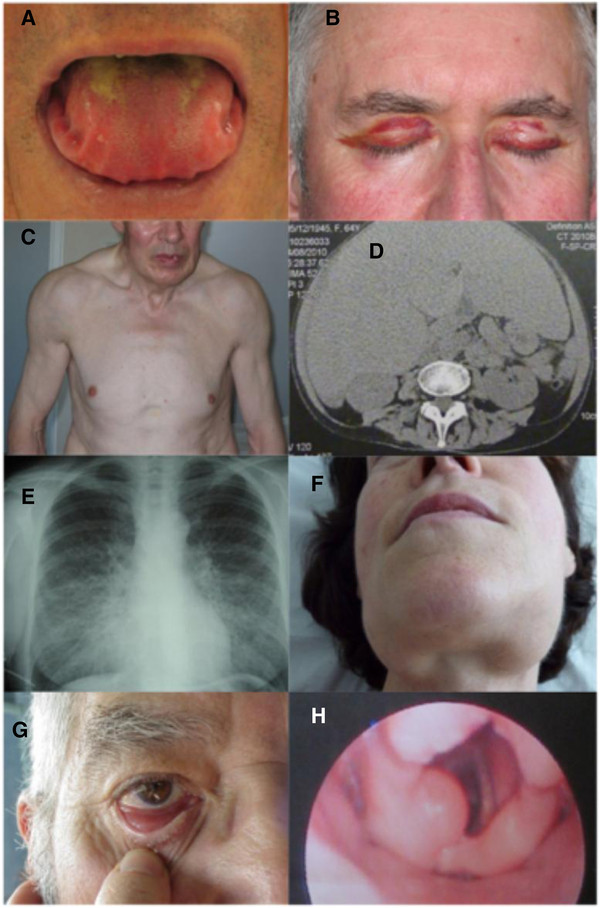
Manifestações sistémicas da amiloidose AL:
A: macroglossia com recorte lateral da língua
B: púrpura periorbital bilateral
C: aspecto pseudo-atlético secundário à infiltração muscular difusa
D: hepatomegalia volumosa devido a amiloidose hepática primária
E: doença pulmonar intersticial bilateral difusa
F: aumento da glândula submandibular
Amiloidose AL localizada:
G: amiloidose conjuntival nodular
H: caroço de amilóide laríngeo supraglótico
História e exame físico:
Biópsia de tecido:
Testes Testes Gonadal Hormones específicos para a doença subjacente:
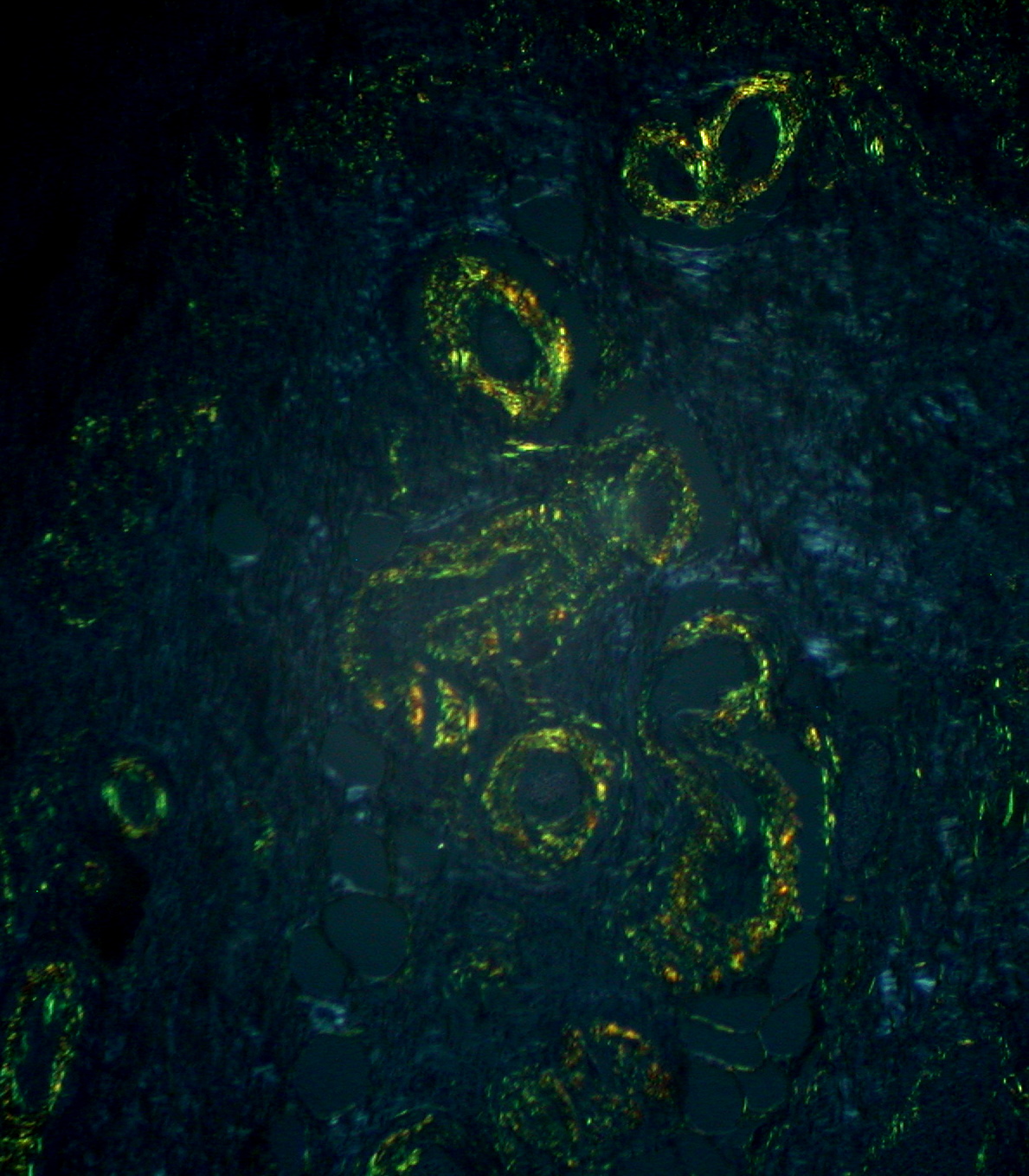
Amiloide: típica birefringência verde maçã com microscopia de luz polarizada após coloração de vermelho do Congo
Imagem: “Gastric Amyloidosis (Congo red stain, crossed polarizers)” por Ed Uthman. Licença: CC BY 2.0
Amiloidose gástrica: deposições de cor vermelha observadas com coloração de H&E em redor dos vasos (contendo eritrócitos)
Imagem: “Gastric Amyloidosis (Congo Red Stain)” por Ed Uthman. Licença: CC BY 2.0