La amiloidosis es una enfermedad causada por el depósito anormal en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum el tejido extracelular de fibrillas compuestas por diversas subunidades proteicas de bajo peso molecular, las cuales se encuentran mal plegadas. Estas proteínas son frecuentemente subproductos de otros procesos patológicos (e.g., el mieloma múltiple). Estas proteínas mal plegadas pueden depositarse en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum diferentes tejidos, interferir con las funciones normales de los LOS Neisseria órganos y causar enfermedades específicas de cada tejido (e.g., la amiloidosis renal causa proteinuria Proteinuria The presence of proteins in the urine, an indicator of kidney diseases. Nephrotic Syndrome in Children). El diagnóstico se establece clínicamente y se confirma con una biopsia de tejido. El tratamiento debe dirigirse a la causa subyacente y a la reducción del depósito de amiloide.
Last updated: Dec 15, 2025
La amiloidosis es una enfermedad causada por el depósito anormal en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum el tejido extracelular de fibrillas compuestas por diversas subunidades proteicas de bajo peso molecular, las cuales se encuentran mal plegadas.
Amiloidosis sistémica:
Amiloidosis localizada:
Amiloidosis hereditaria:
| Enfermedad | Proteína amiloide | Órganos implicados | Detalles específicos |
|---|---|---|---|
| Amiloidosis primaria | AL AL Amyloidosis | Compromiso sistémico:
|
|
| Amiloidosis secundaria | AA AA Amyloidosis | ||
| Amiloidosis asociada a la hemodiálisis | Aβ₂M |
|
| Enfermedad | Proteína amiloide | Órganos implicados | Detalles específicos |
|---|---|---|---|
| Amiloidosis cerebral | Aβ amiloide |
|
|
| Carcinoma medular de tiroides | ACal ACal Amyloidosis | Glándula tiroides | Clínicamente insignificante |
| Diabetes Diabetes Diabetes mellitus (DM) is a metabolic disease characterized by hyperglycemia and dysfunction of the regulation of glucose metabolism by insulin. Type 1 DM is diagnosed mostly in children and young adults as the result of autoimmune destruction of β cells in the pancreas and the resulting lack of insulin. Type 2 DM has a significant association with obesity and is characterized by insulin resistance. Diabetes Mellitus mellitus tipo 2 | AIAPP | Islotes pancreáticos de Langerhans | Asociada al AL Amyloidosis insulinoma Insulinoma A benign tumor of the pancreatic beta cells. Insulinoma secretes excess insulin resulting in hypoglycemia. Pancreatic Neuroendocrine Tumors (PanNETs) |
| Amiloidosis auricular aislada | AANP AANP Amyloidosis | Aurícula | Asociada al AL Amyloidosis envejecimiento |
| Enfermedad | Proteína amiloide | Órganos implicados | Detalles específicos |
|---|---|---|---|
| Fiebre mediterránea familiar | AA AA Amyloidosis |
|
|
| Neuropatía amiloide familiar | ATTR | Nervios periféricos |
|
| Miocardiopatía amiloide familiar | ATTR | Ventrículos |
|
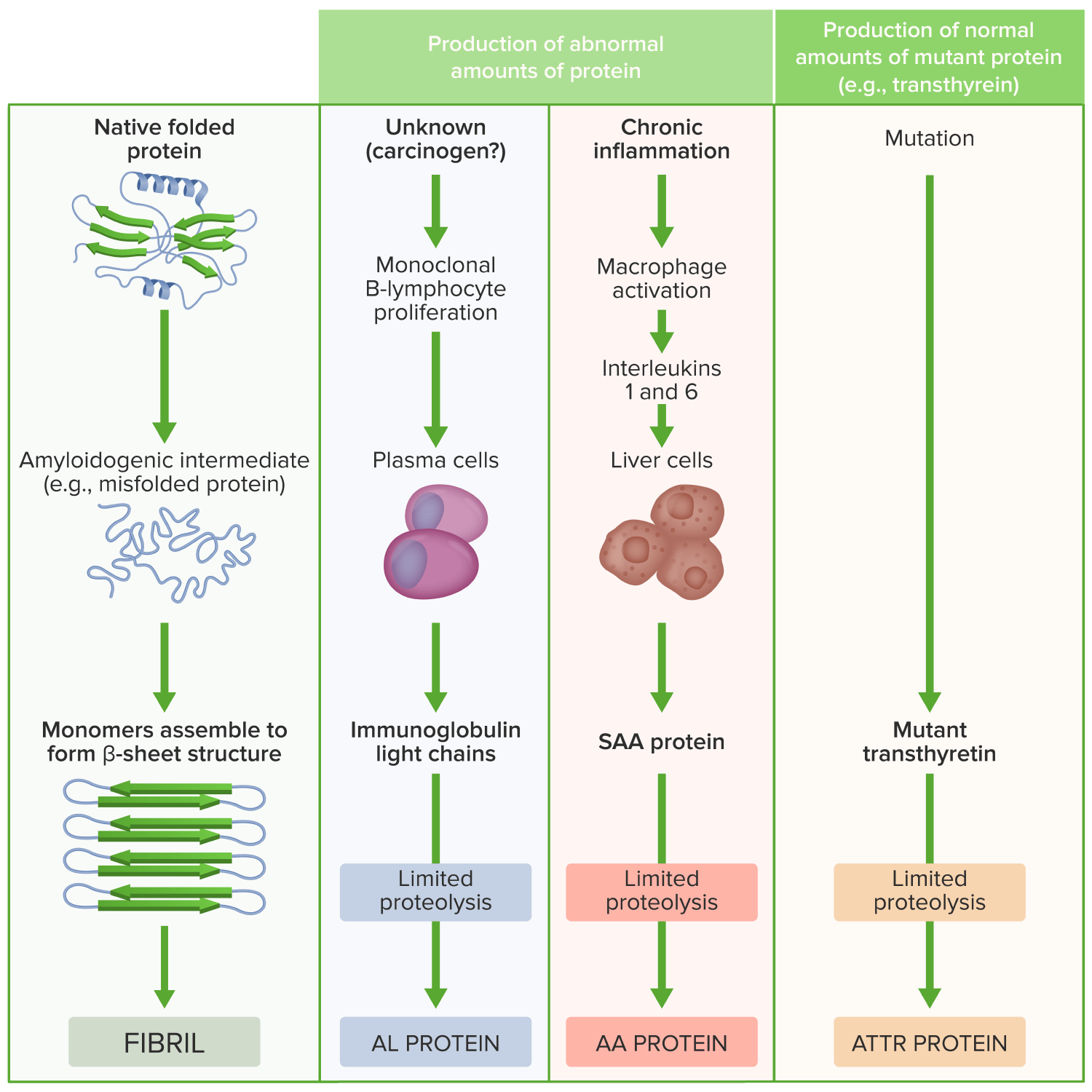
Patogénesis de la amyloidosis
Imagen por Lecturio.La presentación clínica depende del órgano/órganos afectados.
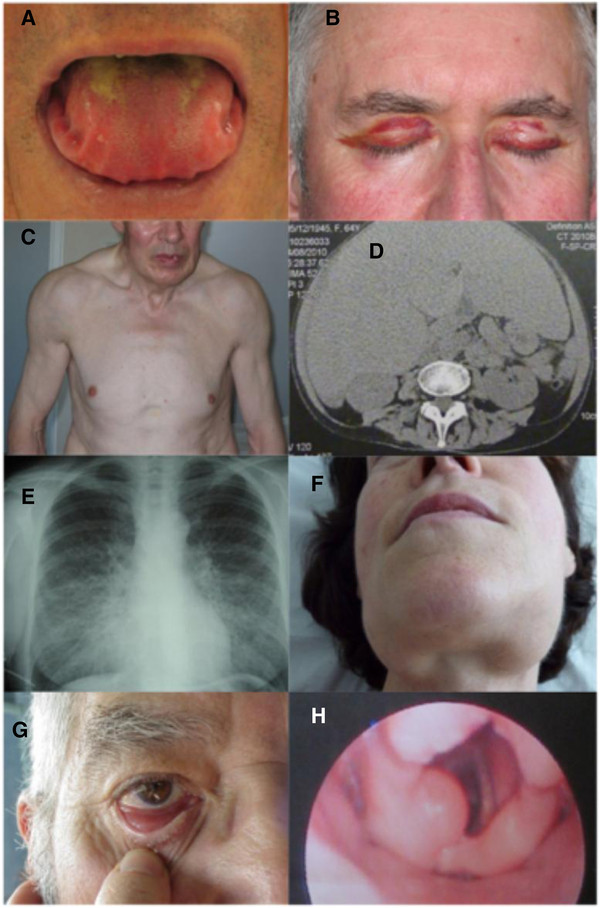
Manifestaciones sistémicas de la amiloidosis AL:
A: macroglosia con festoneado lateral de la lengua
B: púrpura periorbital bilateral
C: aspecto pseudoatletico secundario a una infiltración muscular difusa
D: hepatomegalia voluminosa por amiloidosis hepática primaria
E: enfermedad pulmonar intersticial bilateral difusa
F: agrandamiento de la glándula submandibular
Amiloidosis AL localizada:
G: amiloidosis conjuntival nodular
H: bulto amiloide laríngeo supraglótico
Antecedentes y examen físico:
Biopsia de tejido:
Pruebas específicas de la enfermedad subyacente:
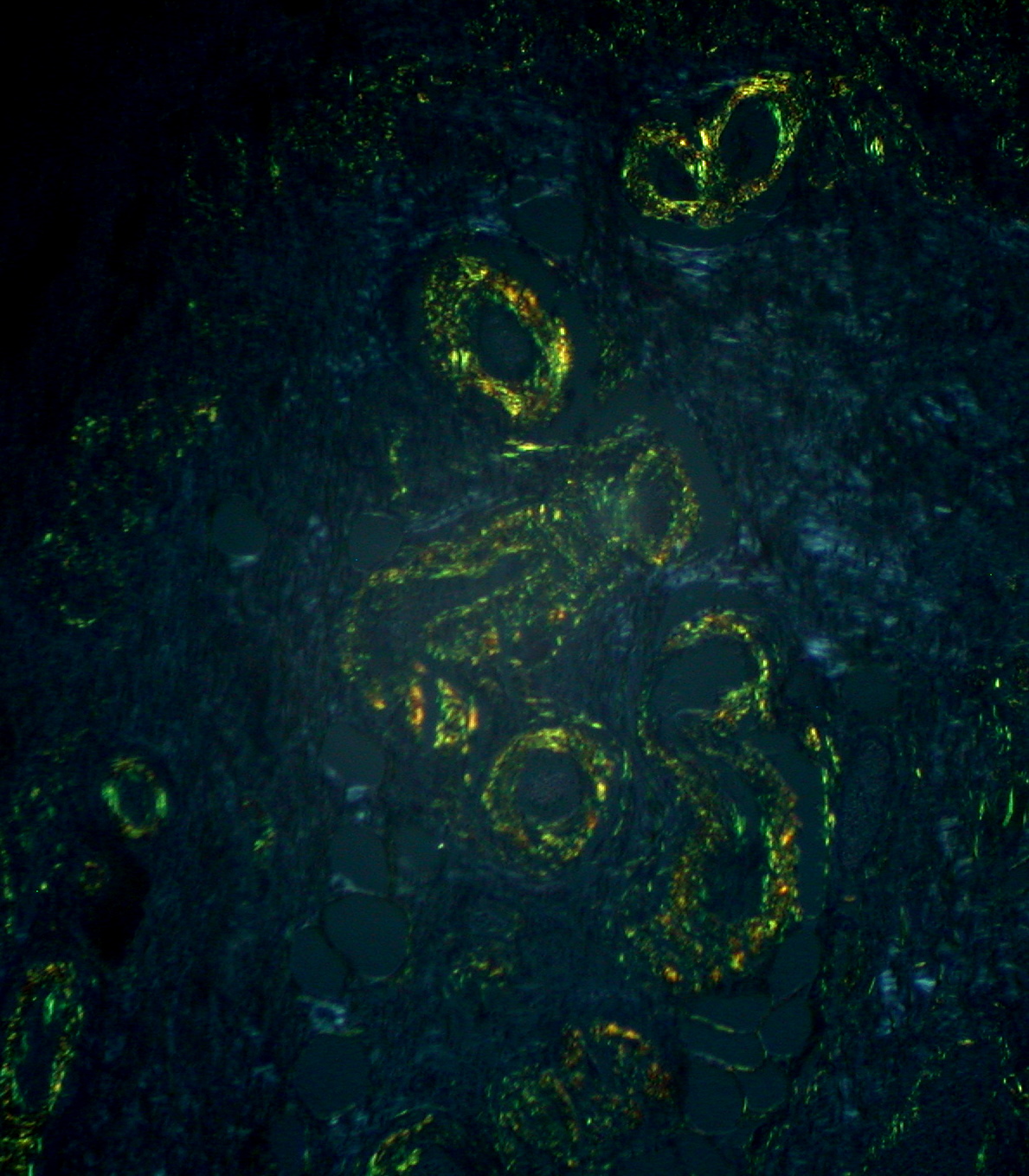
Amiloide: birrefringencia típica de color verde manzana con microscopía de luz polarizada tras la tinción con rojo Congo
Imagen: “Gastric Amyloidosis (Congo red stain, crossed polarizers)” por Ed Uthman. Licencia: CC BY 2.0
Amiloidosis gástrica: depósitos rojos vistos con la tinción H&E alrededor de los vasos (que contienen eritrocitos)
Imagenn: “Gastric Amyloidosis (Congo Red Stain)” por Ed Uthman. Licencia: CC BY 2.0