Gaucher Disease (GD) is an autosomal recessive Autosomal recessive Autosomal inheritance, both dominant and recessive, refers to the transmission of genes from the 22 autosomal chromosomes. Autosomal recessive diseases are only expressed when 2 copies of the recessive allele are inherited. Autosomal Recessive and Autosomal Dominant Inheritance lysosomal storage disorder caused by a deficiency of glucocerebrosidase enzyme activity, resulting in accumulation of glucocerebroside in cells and certain organs. The disease is categorized into 3 types with variable Variable Variables represent information about something that can change. The design of the measurement scales, or of the methods for obtaining information, will determine the data gathered and the characteristics of that data. As a result, a variable can be qualitative or quantitative, and may be further classified into subgroups. Types of Variables clinical presentation. Type 1 Type 1 Spinal Muscular Atrophy is non-neuronopathic, whereas types 2 and 3 are neuronopathic. Manifestations can include bruising, lethargy Lethargy A general state of sluggishness, listless, or uninterested, with being tired, and having difficulty concentrating and doing simple tasks. It may be related to depression or drug addiction. Hyponatremia, anemia Anemia Anemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types, skeletal involvement, and hepatosplenomegaly Hepatosplenomegaly Cytomegalovirus. Neuropathic types can present with cognitive decline, ataxia Ataxia Impairment of the ability to perform smoothly coordinated voluntary movements. This condition may affect the limbs, trunk, eyes, pharynx, larynx, and other structures. Ataxia may result from impaired sensory or motor function. Sensory ataxia may result from posterior column injury or peripheral nerve diseases. Motor ataxia may be associated with cerebellar diseases; cerebral cortex diseases; thalamic diseases; basal ganglia diseases; injury to the red nucleus; and other conditions. Ataxia-telangiectasia, gaze abnormalities, and seizures Seizures A seizure is abnormal electrical activity of the neurons in the cerebral cortex that can manifest in numerous ways depending on the region of the brain affected. Seizures consist of a sudden imbalance that occurs between the excitatory and inhibitory signals in cortical neurons, creating a net excitation. The 2 major classes of seizures are focal and generalized. Seizures. Diagnosis is based on clinical suspicion and confirmed by measurement of glucocerebrosidase activity in peripheral blood leukocytes Leukocytes White blood cells. These include granular leukocytes (basophils; eosinophils; and neutrophils) as well as non-granular leukocytes (lymphocytes and monocytes). White Myeloid Cells: Histology. Genetic analysis can also be used to confirm the diagnosis. Management is supportive and focused on controlling symptoms and improving quality Quality Activities and programs intended to assure or improve the quality of care in either a defined medical setting or a program. The concept includes the assessment or evaluation of the quality of care; identification of problems or shortcomings in the delivery of care; designing activities to overcome these deficiencies; and follow-up monitoring to ensure effectiveness of corrective steps. Quality Measurement and Improvement of life. Enzyme replacement Enzyme replacement Therapeutic replacement or supplementation of defective or missing enzymes to alleviate the effects of enzyme deficiency (e.g., glucosylceramidase replacement for gaucher disease). Severe Combined Immunodeficiency (SCID) therapy, substrate Substrate A substance upon which the enzyme acts. Basics of Enzymes reduction therapy, and bone marrow transplantation Bone marrow transplantation Transfer of hematopoietic stem cells from bone marrow or blood between individuals within the same species (homologous transplantation) or transfer within the same individual (autologous transplantation). Hematopoietic stem cell transplantation has been used as an alternative to bone marrow transplantation in the treatment of a variety of neoplasms. Organ Transplantation (BMT) are treatment options for some patients Patients Individuals participating in the health care system for the purpose of receiving therapeutic, diagnostic, or preventive procedures. Clinician–Patient Relationship.
Last updated: Apr 17, 2025
Gaucher disease (GD) results from a deficiency in the lysosomal hydrolase Lysosomal hydrolase Overview of Lysosomal Storage Diseases beta-glucosidase (glucocerebrosidase).
GD is classified into 3 types:
A deficiency in glucocerebrosidase results in the accumulation of glucocerebroside in reticuloendothelial system cells, which results in:
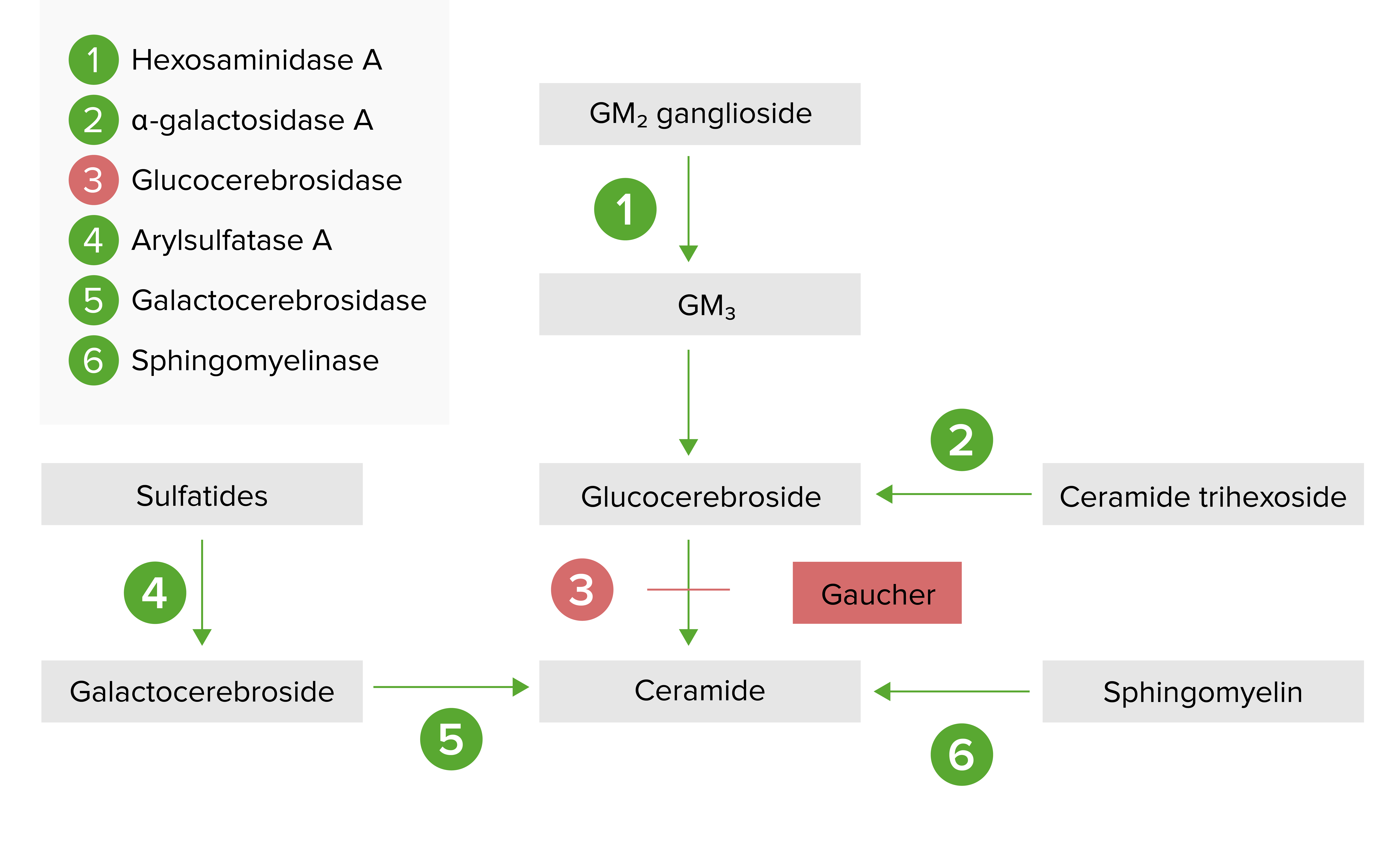
The lysosomal storage pathway:
Gaucher Disease results from deficiency of glucocerebrosidase (step 3), leading to an accumulation of glucocerebroside.
GD is a multisystemic lipidosis with a variable Variable Variables represent information about something that can change. The design of the measurement scales, or of the methods for obtaining information, will determine the data gathered and the characteristics of that data. As a result, a variable can be qualitative or quantitative, and may be further classified into subgroups. Types of Variables clinical presentation.
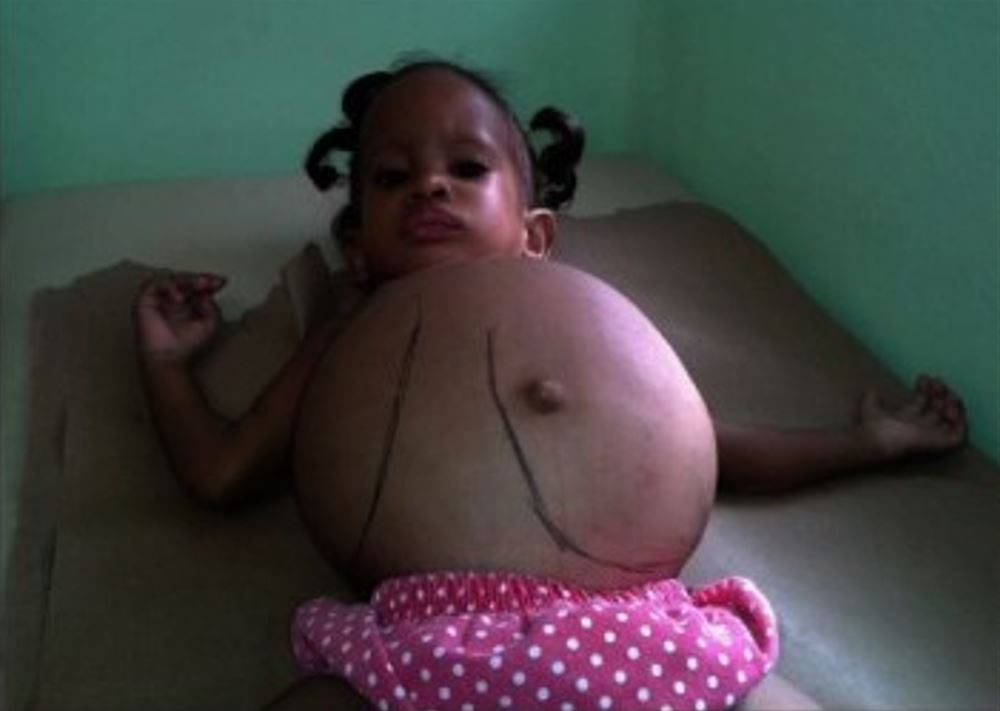
A child with massive hepatosplenomegaly due to Gaucher disease
Image: “Dominican child with Gaucher disease” by Estrada-Veras JI, Cabrera-Peña GA, Pérez-Estrella de Ferrán C. License: CC BY 4.0, cropped by Lecturio.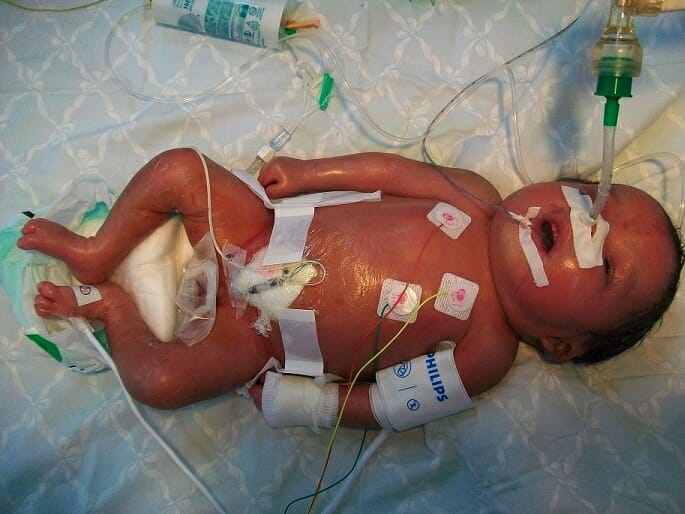
Neonatal presentation of Gaucher disease, type 2 subtype: hydrops, peeling, and shiny skin
Image: “Neonatal presentations of Gaucher disease” by Neonatology Department, Charles Nicolle Hospital, Tunis-El Manar University, Tunis, Tunisia. License: CC BY 2.0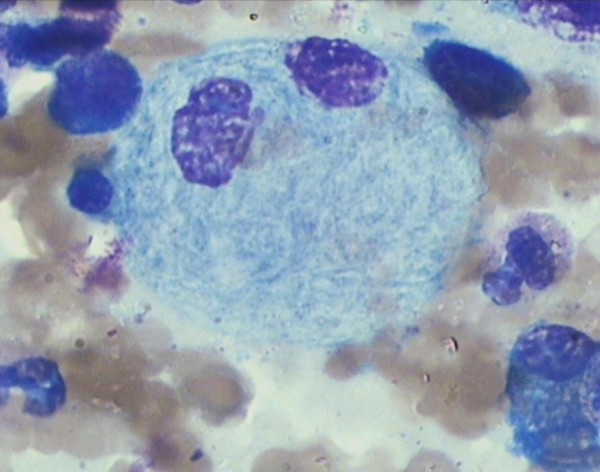
Bone marrow smear showing a Gaucher cell in a patient with type 3 Gaucher disease:
Notice the cytoplasm has a striated, wrinkled appearance.
Treatment is supportive and focused on improving quality Quality Activities and programs intended to assure or improve the quality of care in either a defined medical setting or a program. The concept includes the assessment or evaluation of the quality of care; identification of problems or shortcomings in the delivery of care; designing activities to overcome these deficiencies; and follow-up monitoring to ensure effectiveness of corrective steps. Quality Measurement and Improvement of life. Some patients Patients Individuals participating in the health care system for the purpose of receiving therapeutic, diagnostic, or preventive procedures. Clinician–Patient Relationship with GD have mild symptoms and don’t require treatment.