


A doença de Gaucher (DG) é uma doença autossómica recessiva de armazenamento lisossómico causado por uma deficiência da atividade da enzima glucocerebrosidase, resultando numa acumulação de glucocerebrosídeo nas células e em certos órgãos. A doença está classificada em 3 tipos com apresentações clínicas variáveis. O tipo 1 é não neuronopático, enquanto que os tipos 2 e 3 são neuronopáticos. As manifestações podem incluir hematomas, letargia, anemia Anemia Anemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types, envolvimento esquelético e hepatoesplenomegalia. Os tipos neuropáticos podem apresentar declínio cognitivo, ataxia Ataxia Impairment of the ability to perform smoothly coordinated voluntary movements. This condition may affect the limbs, trunk, eyes, pharynx, larynx, and other structures. Ataxia may result from impaired sensory or motor function. Sensory ataxia may result from posterior column injury or peripheral nerve diseases. Motor ataxia may be associated with cerebellar diseases; cerebral cortex diseases; thalamic diseases; basal ganglia diseases; injury to the red nucleus; and other conditions. Ataxia-telangiectasia, anomalias nos olhos e convulsões. O diagnóstico é baseado na suspeita clínica e confirmado pela medição da atividade da glucocerebrosidase nos leucócitos do sangue periférico. A análise genética também pode ser usada para confirmar o diagnóstico. O tratamento é de apoio e concentra-se no controlo dos sintomas e na melhoria da qualidade de vida. A terapia de reposição enzimática, a terapia de redução do substrato e o transplante de medula óssea (TMO) são opções de tratamento para alguns pacientes.
Last updated: Apr 17, 2025
A doença de Gaucher (DG) resulta de uma deficiência na beta-glucosidase Beta-glucosidase An exocellulase with specificity for a variety of beta-d-glucoside substrates. It catalyzes the hydrolysis of terminal non-reducing residues in beta-d-glucosides with release of glucose. Gaucher Disease hidrolase (glucocerebrosidase) lisossomal.
A DG é classificado em 3 tipos:
Uma deficiência em glucocerebrosidase resulta na acumulação de glucocerebrosídeo nas células do sistema reticuloendotelial, o que resulta em:
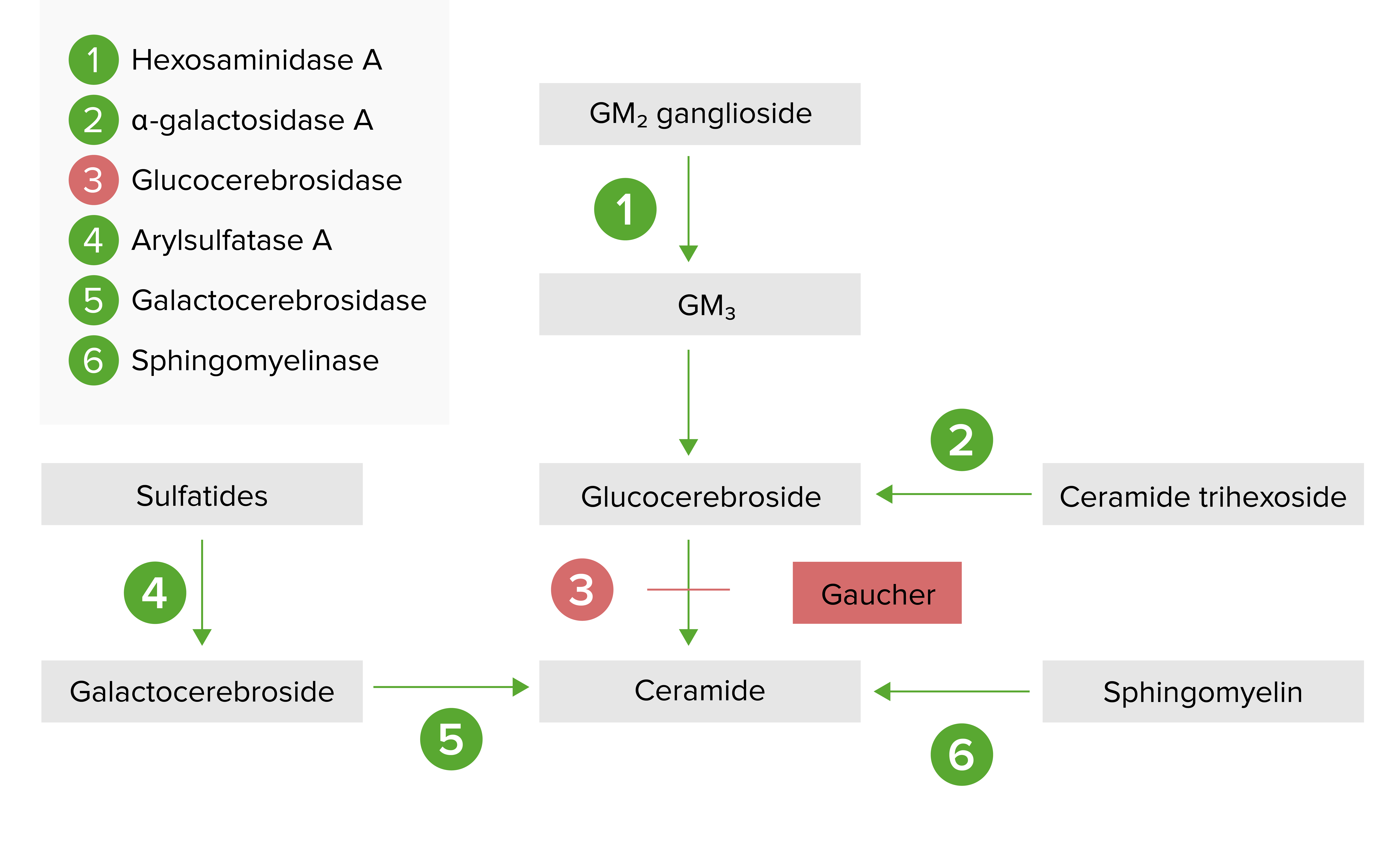
A via de armazenamento lisossomal:
A doença de Gaucher resulta da deficiência de glucocerebrosidase (passo 3), levando a uma acumulação de glucocerebrosídeo.
A DG é uma lipidose multisistémica com uma apresentação clínica variável.
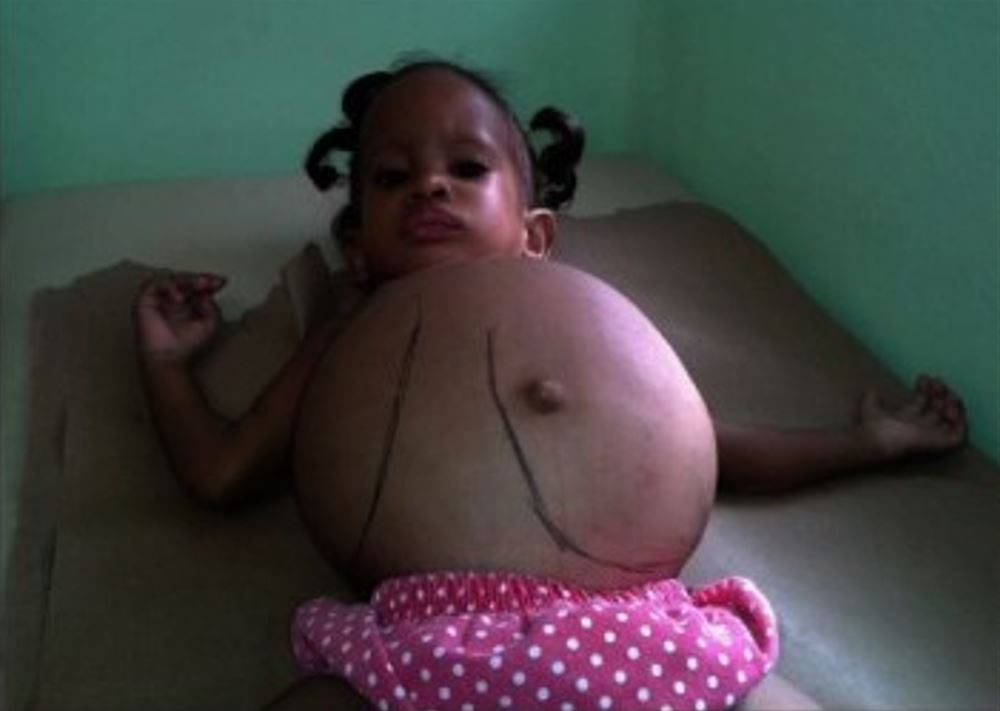
Uma criança com hepatoesplenomegalia acentuada devido à doença de Gaucher
Imagem: “Dominican child with Gaucher disease” by Estrada-Veras JI, Cabrera-Peña GA, Pérez-Estrella de Ferrán C. License: CC BY 4.0, cropped by Lecturio.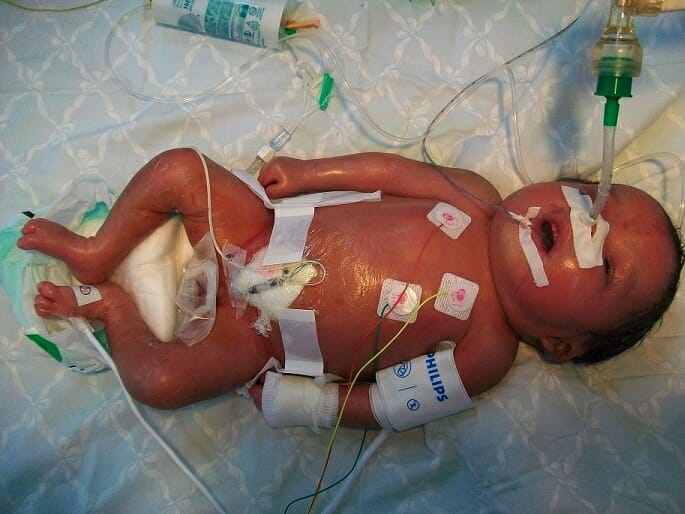
Apresentação neonatal da doença de Gaucher, tipo 2: hidrópsia, descamação e pele brilhante
Imagem: “Neonatal presentations of Gaucher disease” por Neonatology Department, Charles Nicolle Hospital, Tunis-El Manar University, Tunis, Tunisia. Licença: CC BY 2.0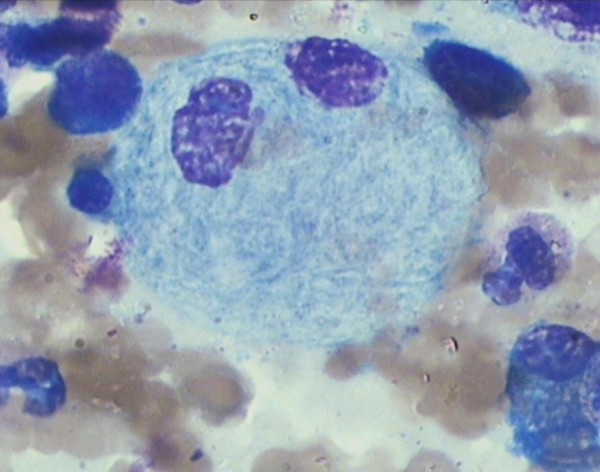
Esfregaço de medula óssea que mostra uma célula Gaucher num paciente com doença de Gaucher tipo 3:
Note-se que o citoplasma tem uma aparência estriada e enrugada.
O tratamento é de apoio e focado na melhoria da qualidade de vida. Alguns pacientes com DG têm sintomas leves e não precisam de tratamento.
