Tay-Sachs disease is an autosomal recessive Autosomal recessive Autosomal inheritance, both dominant and recessive, refers to the transmission of genes from the 22 autosomal chromosomes. Autosomal recessive diseases are only expressed when 2 copies of the recessive allele are inherited. Autosomal Recessive and Autosomal Dominant Inheritance lysosomal storage disorder caused by genetic mutations Genetic Mutations Carcinogenesis in the hexosaminidase A (HEXA) gene Gene A category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms. Basic Terms of Genetics, leading to progressive neurodegeneration. This disease is highly prevalent in the Ashkenazi Jewish population. Classic symptoms in infants include rapid degeneration of cognitive and neuromuscular abilities, progressive blindness Blindness The inability to see or the loss or absence of perception of visual stimuli. This condition may be the result of eye diseases; optic nerve diseases; optic chiasm diseases; or brain diseases affecting the visual pathways or occipital lobe. Retinopathy of Prematurity, and a macular cherry-red spot on physical examination. Diagnosis is made by measurement of hexosaminidase A and beta-hexosaminidase enzyme activity. Currently, there is no cure for this disease, and treatment is aimed at improving the patient’s quality Quality Activities and programs intended to assure or improve the quality of care in either a defined medical setting or a program. The concept includes the assessment or evaluation of the quality of care; identification of problems or shortcomings in the delivery of care; designing activities to overcome these deficiencies; and follow-up monitoring to ensure effectiveness of corrective steps. Quality Measurement and Improvement of life.
Last updated: Apr 23, 2025
Tay-Sachs disease is a lysosomal storage disorder caused by a deficiency in the hexosaminidase A (Hex-A) enzyme.
Tay-Sachs disease is classified into 3 variants based on age of onset and enzyme activity.
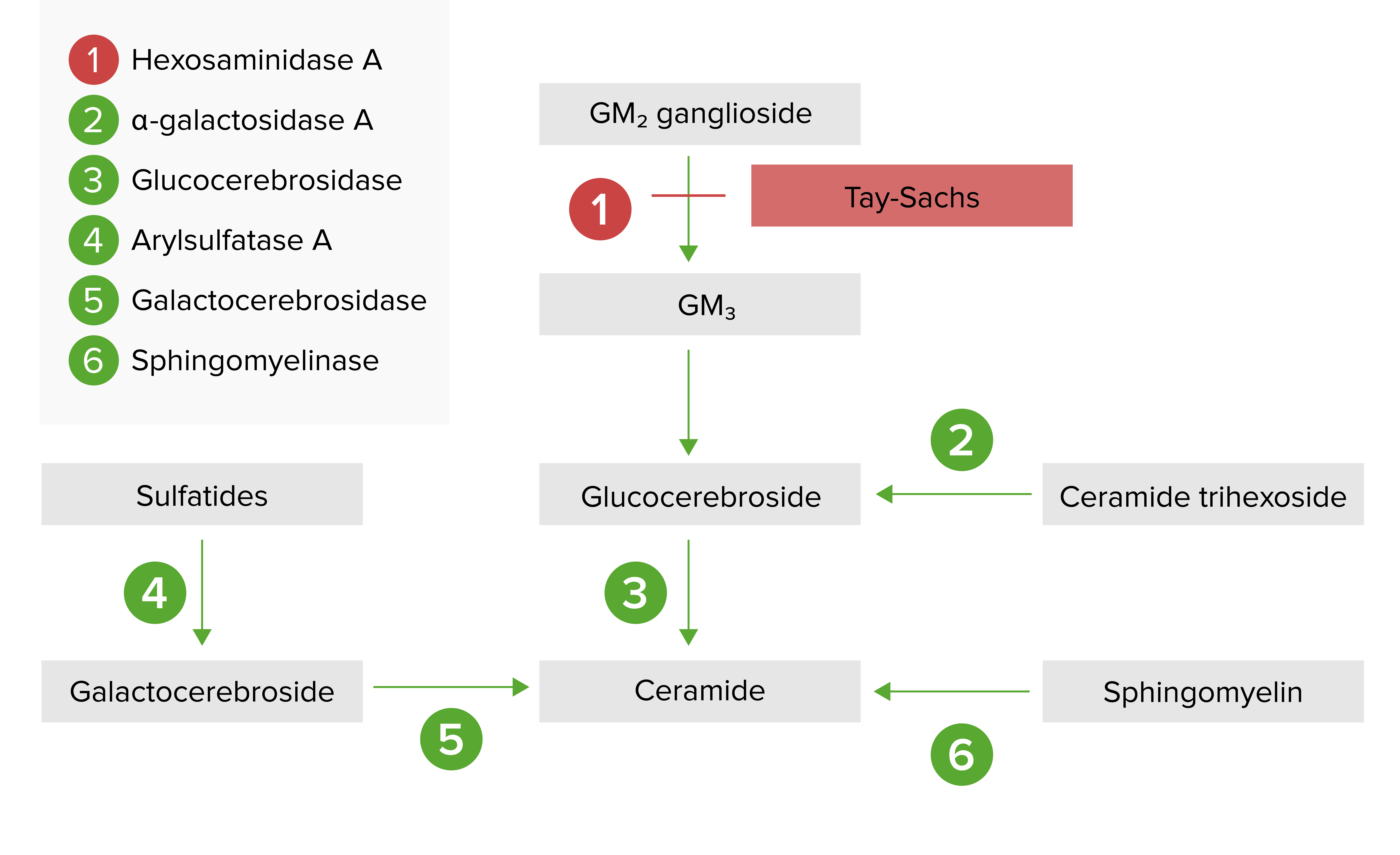
The lysosomal storage pathway:
Tay-Sachs disease results from hexosaminidase A deficiency, resulting in a build-up of GM2 ganglioside.
Tay-Sachs disease is a neurodegenerative disorder with a variable Variable Variables represent information about something that can change. The design of the measurement scales, or of the methods for obtaining information, will determine the data gathered and the characteristics of that data. As a result, a variable can be qualitative or quantitative, and may be further classified into subgroups. Types of Variables clinical presentation depending on the variant.
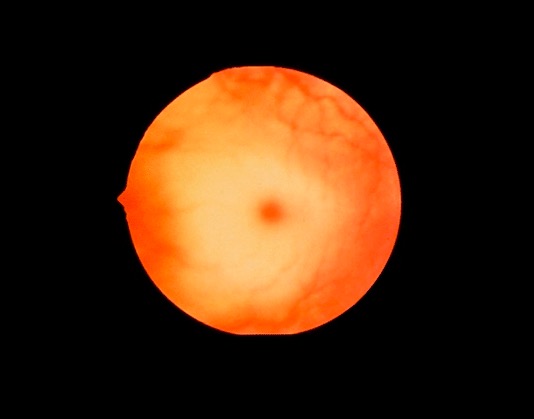
Cherry-red spot:
The macula is normally red. Pallor develops around the red macula due to glycolipid accumulation, giving the appearance of a cherry-red spot. The spot usually develops by age 1 in Tay-Sachs disease, and can also be found in other lysosomal storage disorders.
Testing is pursued following clinical suspicion. Prenatal screening Screening Preoperative Care is also possible via chorionic villus sampling or amniocentesis Amniocentesis Percutaneous transabdominal puncture of the uterus during pregnancy to obtain amniotic fluid. It is commonly used for fetal karyotype determination in order to diagnose abnormal fetal conditions. Polyhydramnios.
There is no cure for Tay-Sachs disease. Management is supportive, focused on controlling symptoms and improving quality Quality Activities and programs intended to assure or improve the quality of care in either a defined medical setting or a program. The concept includes the assessment or evaluation of the quality of care; identification of problems or shortcomings in the delivery of care; designing activities to overcome these deficiencies; and follow-up monitoring to ensure effectiveness of corrective steps. Quality Measurement and Improvement of life.