Niemann-Pick disease (NPD), also known as sphingomyelinase deficiency, is a rare, inherited, lysosomal storage disorder. The disease is classified on the basis of the genetic mutation Mutation Genetic mutations are errors in DNA that can cause protein misfolding and dysfunction. There are various types of mutations, including chromosomal, point, frameshift, and expansion mutations. Types of Mutations. Type A and type B result from mutations in the SMPD-1 gene Gene A category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms. Basic Terms of Genetics, resulting in acid sphingomyelinase enzyme deficiency. Type C results from NPC1 or NPC2 gene Gene A category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms. Basic Terms of Genetics mutations, which are needed for intracellular transport of lipids Lipids Lipids are a diverse group of hydrophobic organic molecules, which include fats, oils, sterols, and waxes. Fatty Acids and Lipids. These mutations result in lysosomes Lysosomes A class of morphologically heterogeneous cytoplasmic particles in animal and plant tissues characterized by their content of hydrolytic enzymes and the structure-linked latency of these enzymes. The intracellular functions of lysosomes depend on their lytic potential. The single unit membrane of the lysosome acts as a barrier between the enzymes enclosed in the lysosome and the external substrate. The activity of the enzymes contained in lysosomes is limited or nil unless the vesicle in which they are enclosed is ruptured or undergoes membrane fusion. The Cell: Organelles that are unable to properly metabolize fat, such as sphingomyelin Sphingomyelin A class of sphingolipids found largely in the brain and other nervous tissue. They contain phosphocholine or phosphoethanolamine as their polar head group so therefore are the only sphingolipids classified as phospholipids. Neonatal Respiratory Distress Syndrome and cholesterol Cholesterol The principal sterol of all higher animals, distributed in body tissues, especially the brain and spinal cord, and in animal fats and oils. Cholesterol Metabolism, resulting in progressive intracellular lipid accumulation and organ damage. Clinical manifestations may include failure to thrive Failure to Thrive Failure to thrive (FTT), or faltering growth, describes suboptimal weight gain and growth in children. The majority of cases are due to inadequate caloric intake; however, genetic, infectious, and oncological etiologies are also common. Failure to Thrive, hepatosplenomegaly Hepatosplenomegaly Cytomegalovirus, thrombocytopenia Thrombocytopenia Thrombocytopenia occurs when the platelet count is < 150,000 per microliter. The normal range for platelets is usually 150,000-450,000/µL of whole blood. Thrombocytopenia can be a result of decreased production, increased destruction, or splenic sequestration of platelets. Patients are often asymptomatic until platelet counts are < 50,000/µL. Thrombocytopenia, interstitial lung disease, cognitive and motor Motor Neurons which send impulses peripherally to activate muscles or secretory cells. Nervous System: Histology impairment, and macular cherry-red spots. Progressive neurodegeneration and a short life expectancy Life expectancy Based on known statistical data, the number of years which any person of a given age may reasonably expected to live. Population Pyramids are seen in NPD-A, while NPD-B is typically nonneurogenic. Diagnosis of NPD is based on clinical suspicion and can be confirmed by the measurement of sphingomyelinase activity or biomarkers, genetic testing Genetic Testing Detection of a mutation; genotype; karyotype; or specific alleles associated with genetic traits, heritable diseases, or predisposition to a disease, or that may lead to the disease in descendants. It includes prenatal genetic testing. Myotonic Dystrophies, or biopsy Biopsy Removal and pathologic examination of specimens from the living body. Ewing Sarcoma. There is currently no cure for NPD, so management is supportive and focuses on controlling symptoms.
Last updated: Dec 15, 2025
Niemann-Pick disease (NPD) is an autosomal recessive Autosomal recessive Autosomal inheritance, both dominant and recessive, refers to the transmission of genes from the 22 autosomal chromosomes. Autosomal recessive diseases are only expressed when 2 copies of the recessive allele are inherited. Autosomal Recessive and Autosomal Dominant Inheritance lysosomal storage disorder that is classified on the basis of the gene mutation Gene Mutation Myotonic Dystrophies and enzyme deficiency:
Normal lysosomes Lysosomes A class of morphologically heterogeneous cytoplasmic particles in animal and plant tissues characterized by their content of hydrolytic enzymes and the structure-linked latency of these enzymes. The intracellular functions of lysosomes depend on their lytic potential. The single unit membrane of the lysosome acts as a barrier between the enzymes enclosed in the lysosome and the external substrate. The activity of the enzymes contained in lysosomes is limited or nil unless the vesicle in which they are enclosed is ruptured or undergoes membrane fusion. The Cell: Organelles:
In NPD-A and B:
In NPD-C:
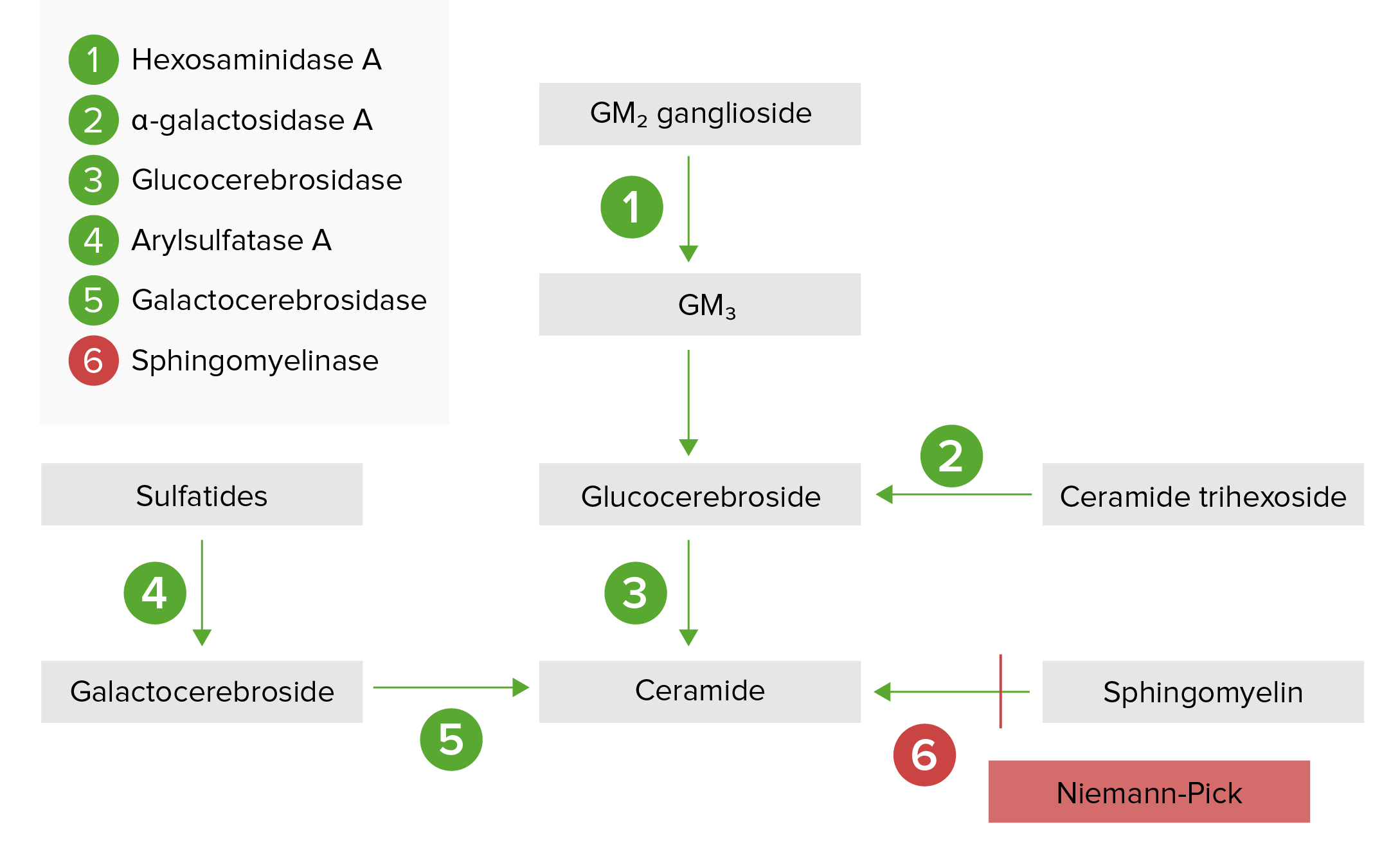
The lysosomal storage pathway:
Niemann-Pick disease (types A and B) results from acid sphingomyelinase deficiency, resulting in a buildup of sphingomyelin.
The clinical presentation of NPD depends on the type and severity of the disease.
Disease course:
Signs and symptoms:
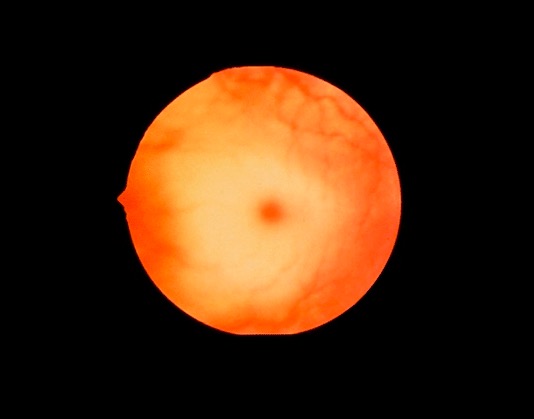
Fundoscopic image demonstrating a cherry-red spot, which may be seen in Niemann-Pick disease
Image: “Cherry red spot” by Jonathan Trobe, M.D. License: CC BY 3.0Disease course:
Signs and symptoms:
Disease course:
Signs and symptoms:
Suspicion for NPD is raised on the basis of the clinical features. The diagnosis can be confirmed with the following investigations:
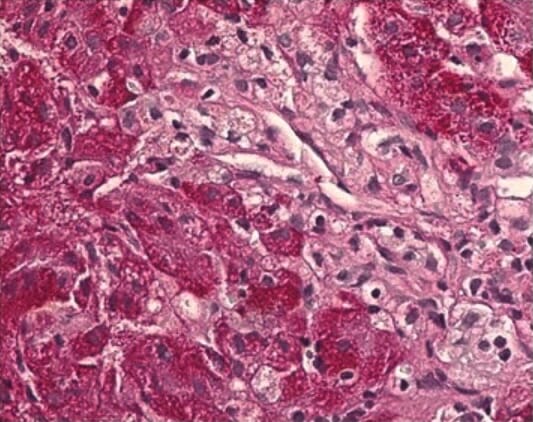
Histopathological liver biopsy showing swollen Kupffer cells with a foamy cytoplasm, typical for NPD-C
Image: “Histopathological liver biopsy findings” by Federal State Budget Institution, Research Center for Obstetrics, Gynecology and Perinatology, Federal State Budget Institution, 117997 Oparina str. 4, Moscow, Russia. License: CC BY 4.0There is no cure for NPD. Management is supportive and multidisciplinary.