Las proteínas son 1 de los LOS Neisseria 3 macronutrientes primarios en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum el cuerpo y se sintetizan a partir de bloques de construcción individuales llamados aminoácidos. Los LOS Neisseria aminoácidos se unen mediante enlaces peptídicos, que unen el extremo amino de un aminoácido con el extremo carboxi del siguiente aminoácido, generando la estructura primaria de una proteína. Luego, la hebra del aminoácido sufre un plegamiento adicional, generando finalmente estructuras 3-dimensionales complejas. Las proteínas tienen una amplia variedad de funciones, incluidas funciones catalíticas, estructurales, reguladoras, de transporte, almacenamiento e inmunológicas. Son digeridas por proteasas y peptidasas secretadas por el estómago y el páncreas y absorbidas como aminoácidos individuales en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum el intestino delgado a través de transportadores especializados. Existen innumerables afecciones médicas relacionadas con anomalías proteicas, incluidas anomalías relacionadas con enzimas, receptores, canales de membrana, hormonas, acumulación de proteínas y trastornos autoinmunes.
Last updated: Dec 15, 2025
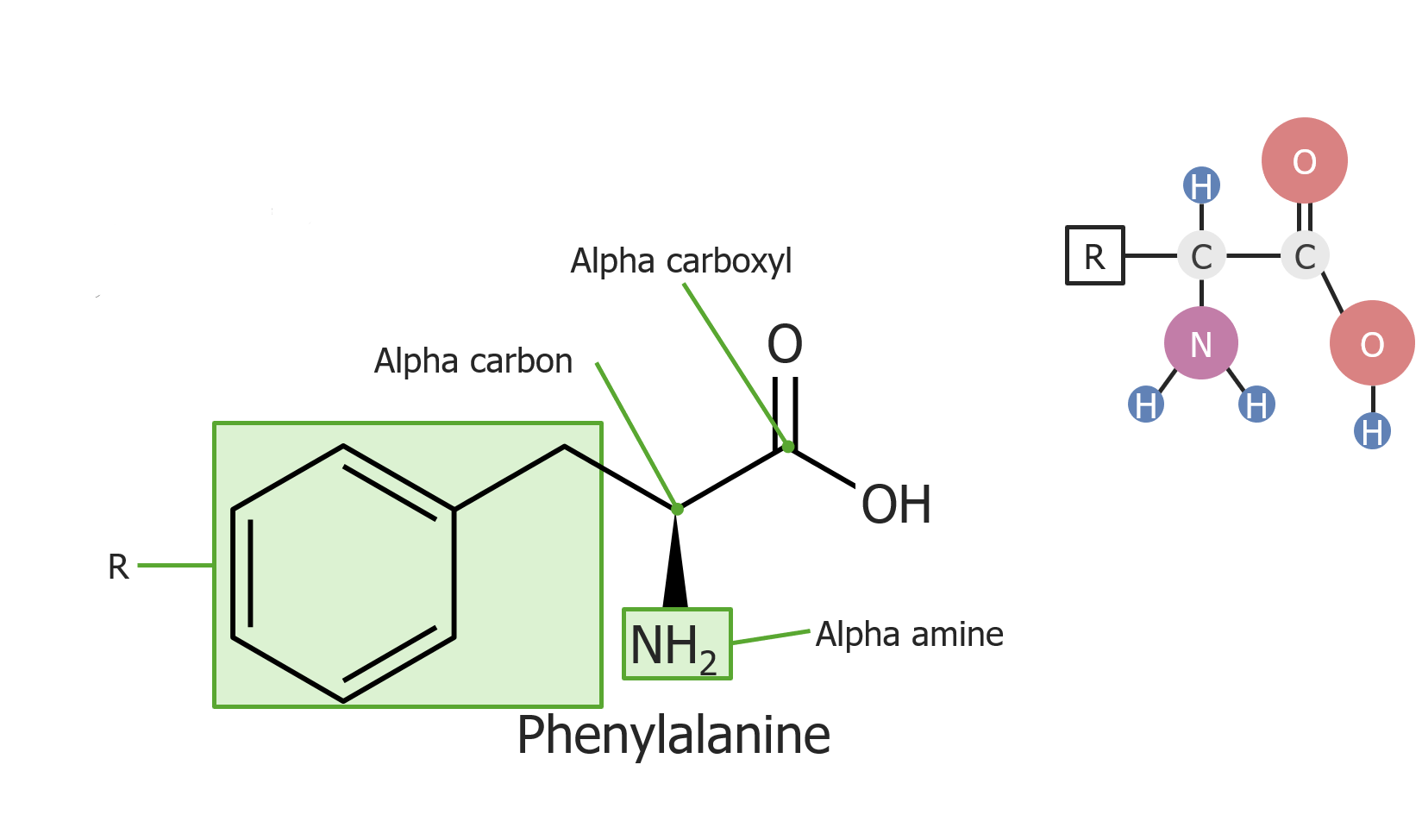
Ejemplo del aminoácido fenilalanina
Imagen por Lecturio.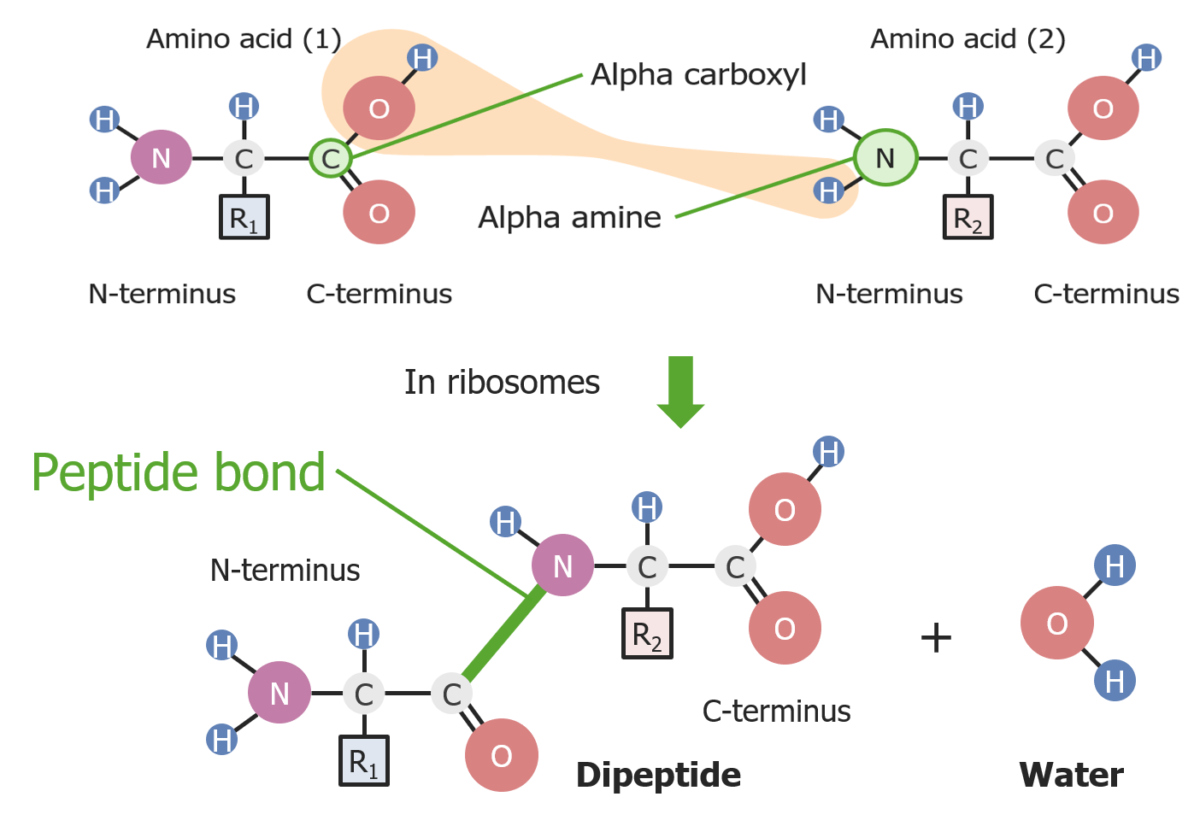
Formación de un enlace peptídico entre 2 aminoácidos
Imagen por Lecturio.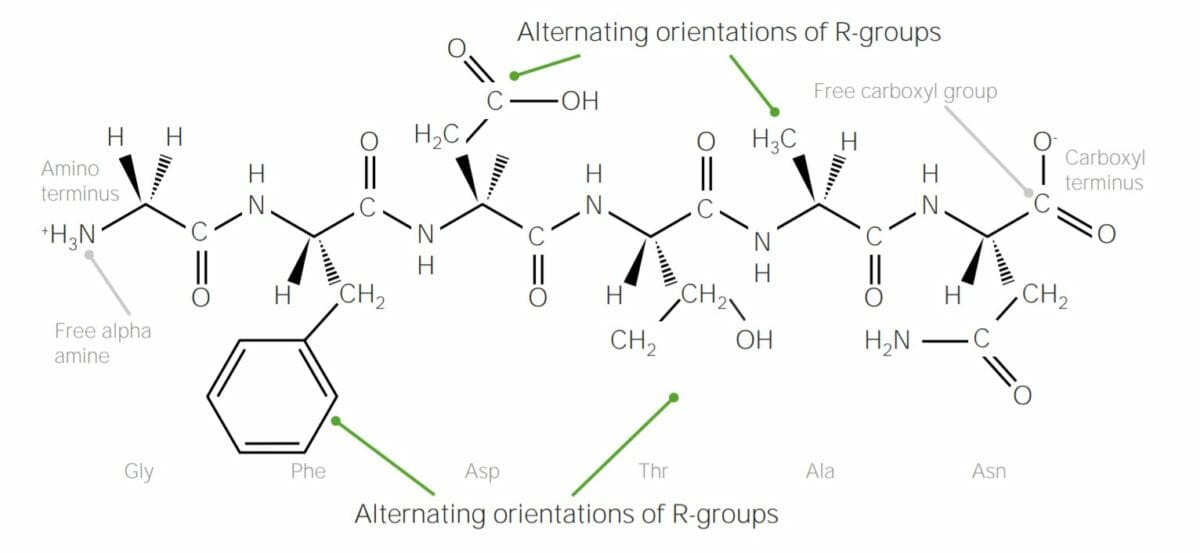
Ejemplo de polipéptido que contiene 6 aminoácidos, unidos entre sí mediante enlaces peptídicos:
Obsérvese cómo los grupos R de cada aminoácido alternan los “lados” de la cadena polipeptídica; esto se debe a la configuración trans de los enlaces peptídicos.
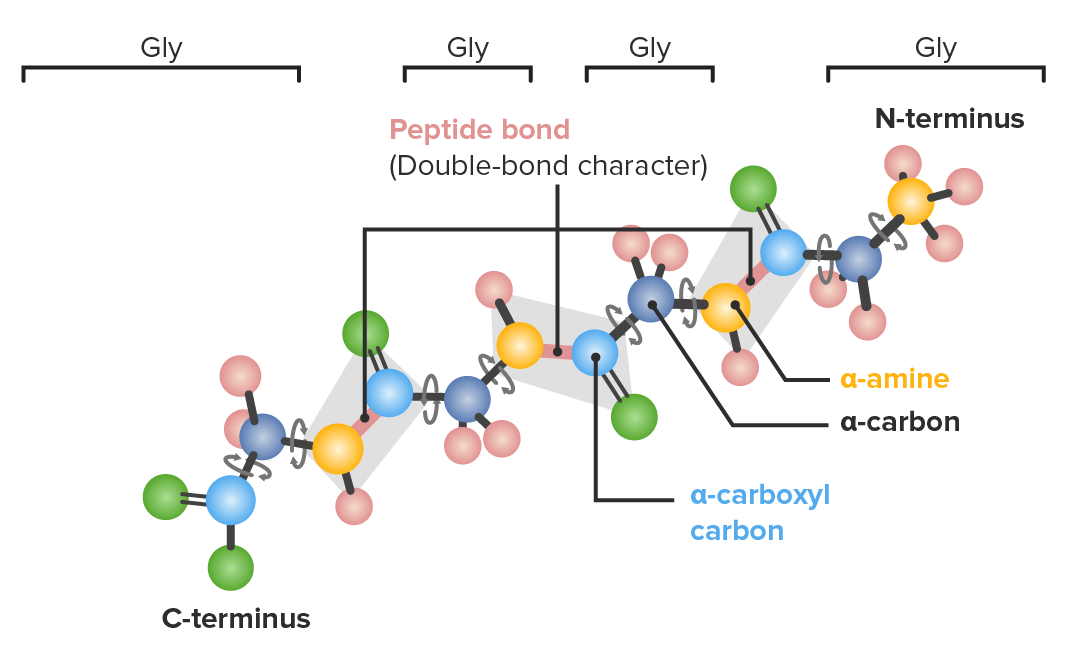
Ejemplo de un polipéptido con 4 aminoácidos de glicina (gly) en secuencia que demuestra los enlaces que tienen libertad para rotar:
Azul oscuro: carbonos α
Azul claro: carbonos carboxilo
Amarillo: nitrógeno
Verde: oxígeno
Rosa: hidrógeno
Existen 4 niveles de estructuras proteicas; estos a menudo se denominan plegamiento de proteínas. Los LOS Neisseria niveles estructurales son, estructuras primarias, secundarias, terciarias y cuaternarias. El plegamiento adecuado requiere la asistencia de proteínas chaperonas.
Estructura primaria:
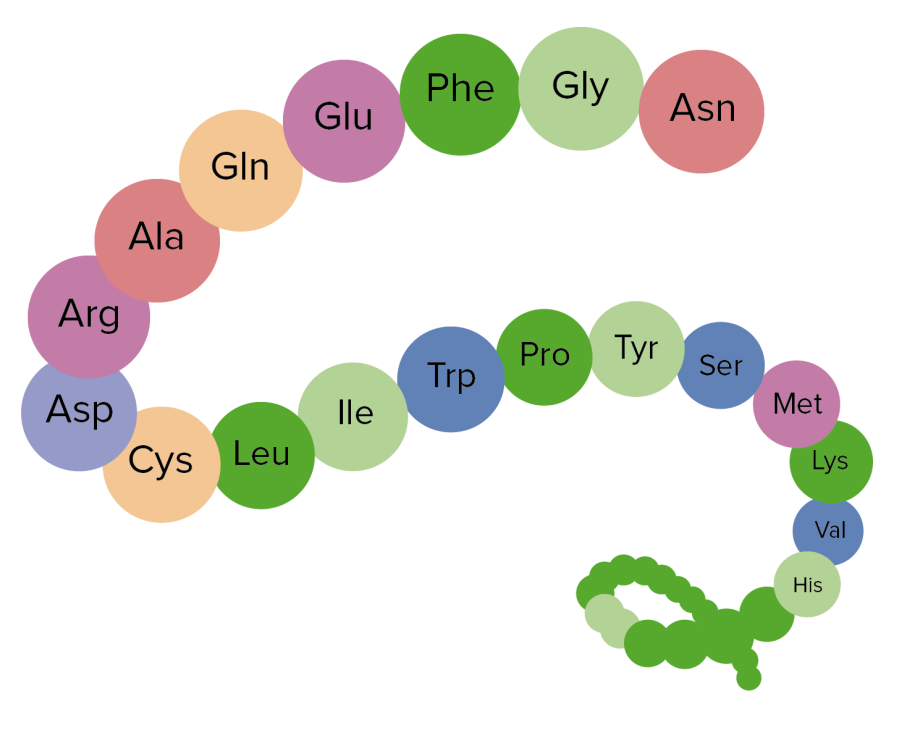
Imagen que muestra la estructura primaria de las proteínas, una agregación de aminoácidos
Imagen por Lecturio.Estructura secundaria:
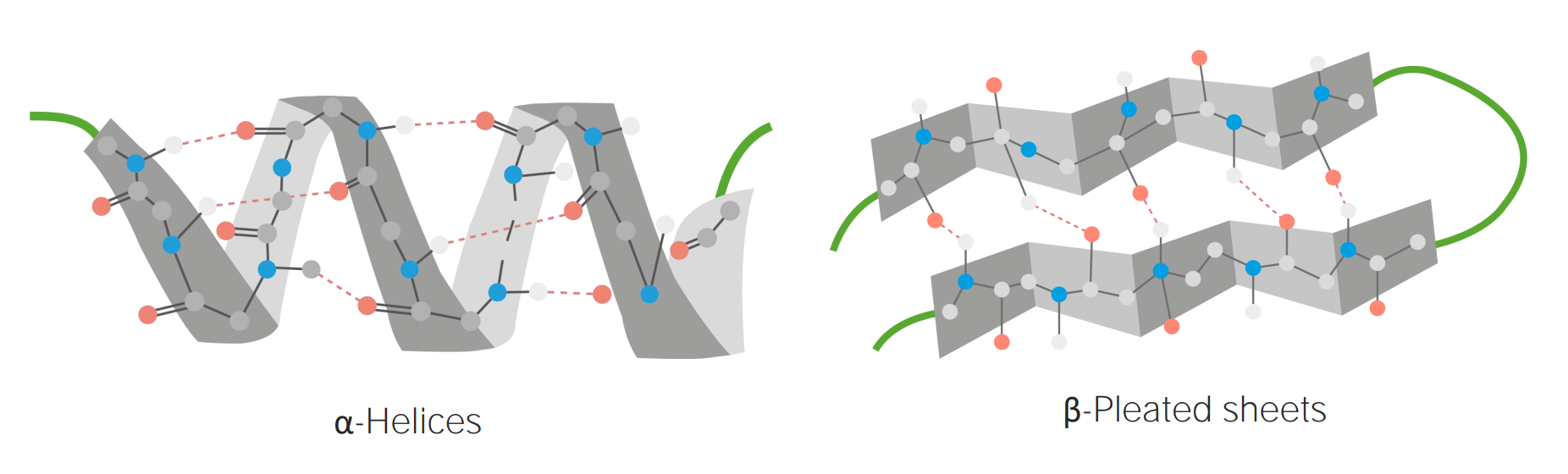
Ejemplos de α-hélice y β-laminar plegada
Imagen por Lecturio.Estructura terciaria:
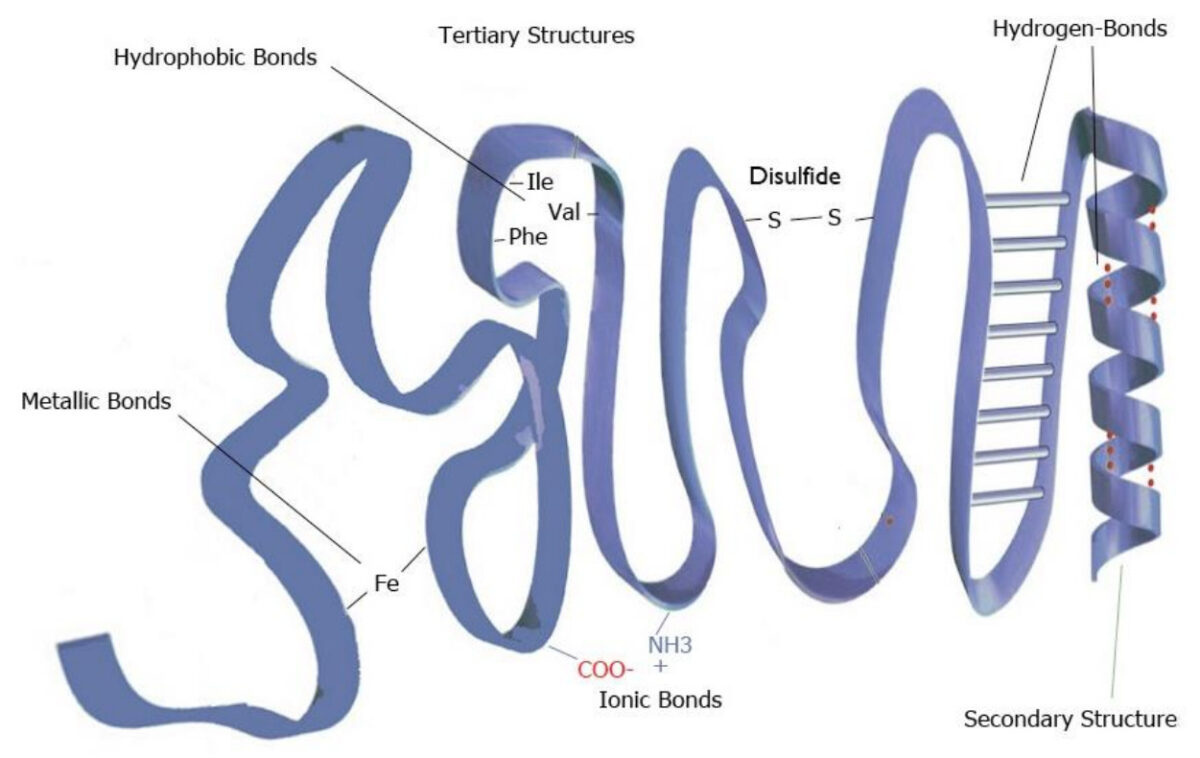
Ejemplo de estructura terciaria
Imagen por Lecturio.
Imagen que representa la estructura terciaria de las proteínas
Imagen por Lecturio.Estructura cuaternaria:
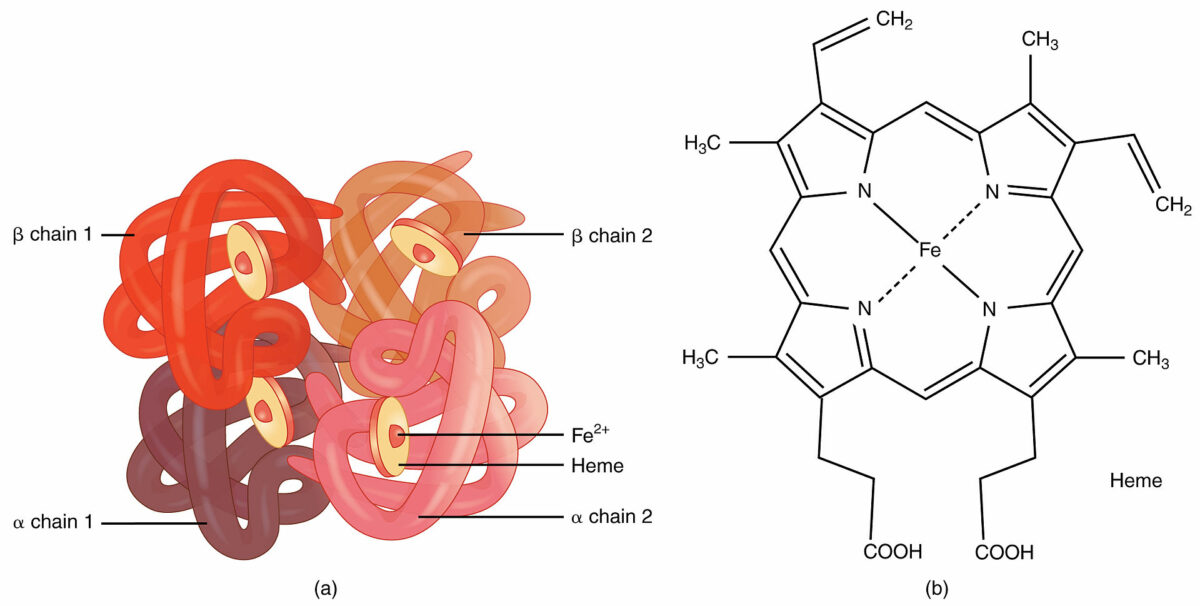
Hemoglobina:
Un ejemplo de estructura cuaternaria
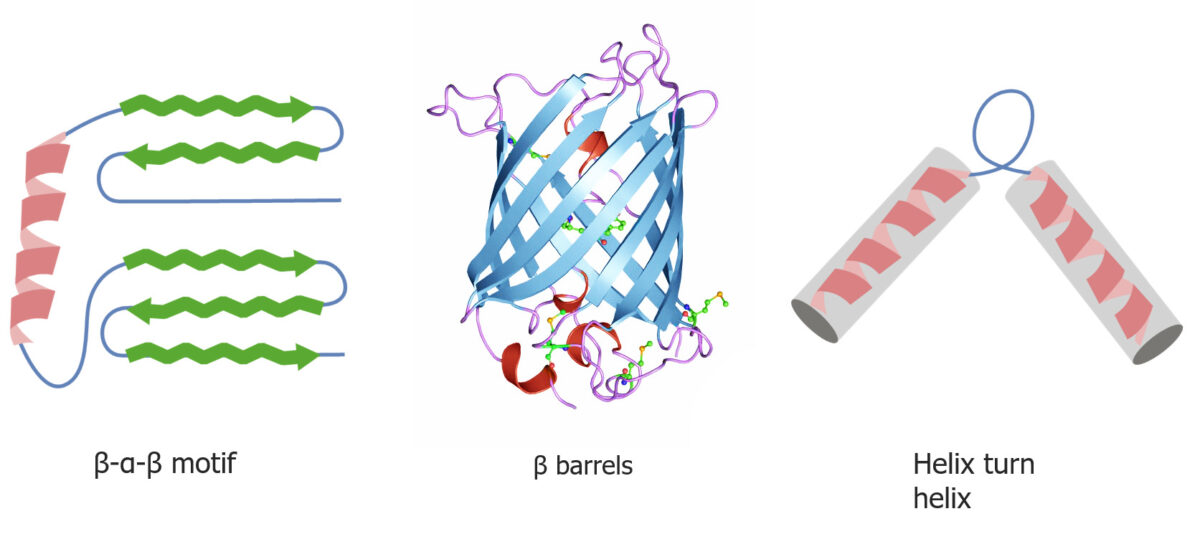
Motivos de plegamiento de proteínas cuaternarias y terciarias
Imagen por Lecturio.Las proteínas chaperonas ayudan en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum el plegamiento de las proteínas.
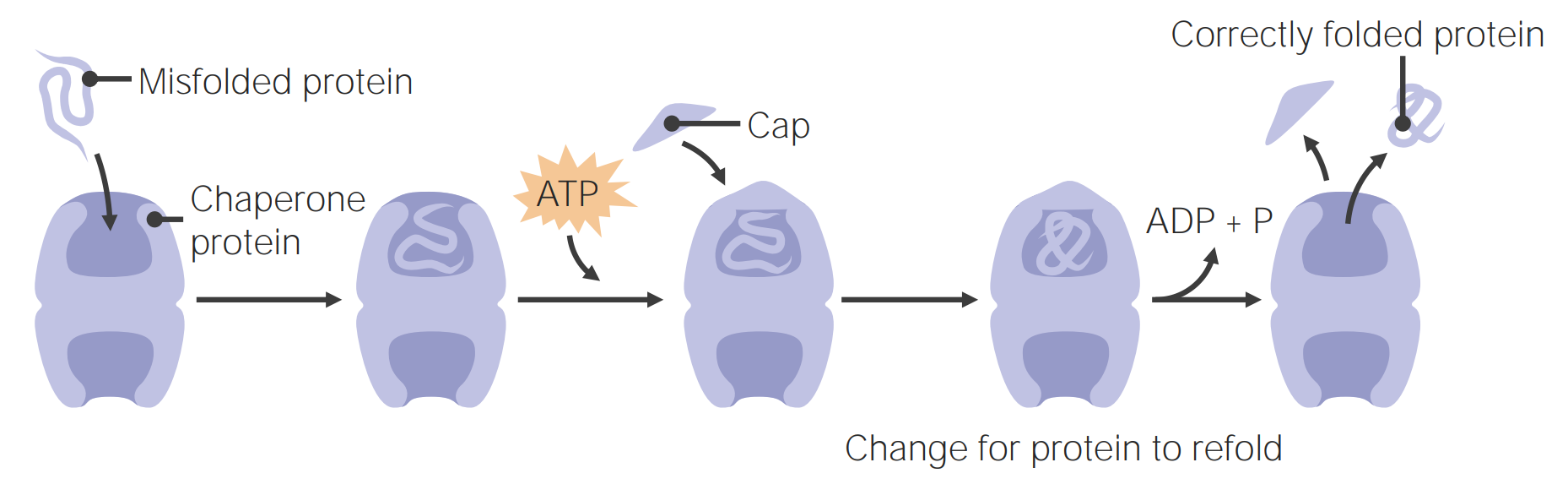
Las proteínas chaperonas ayudan en el plegamiento de las proteínas
Imagen por Lecturio.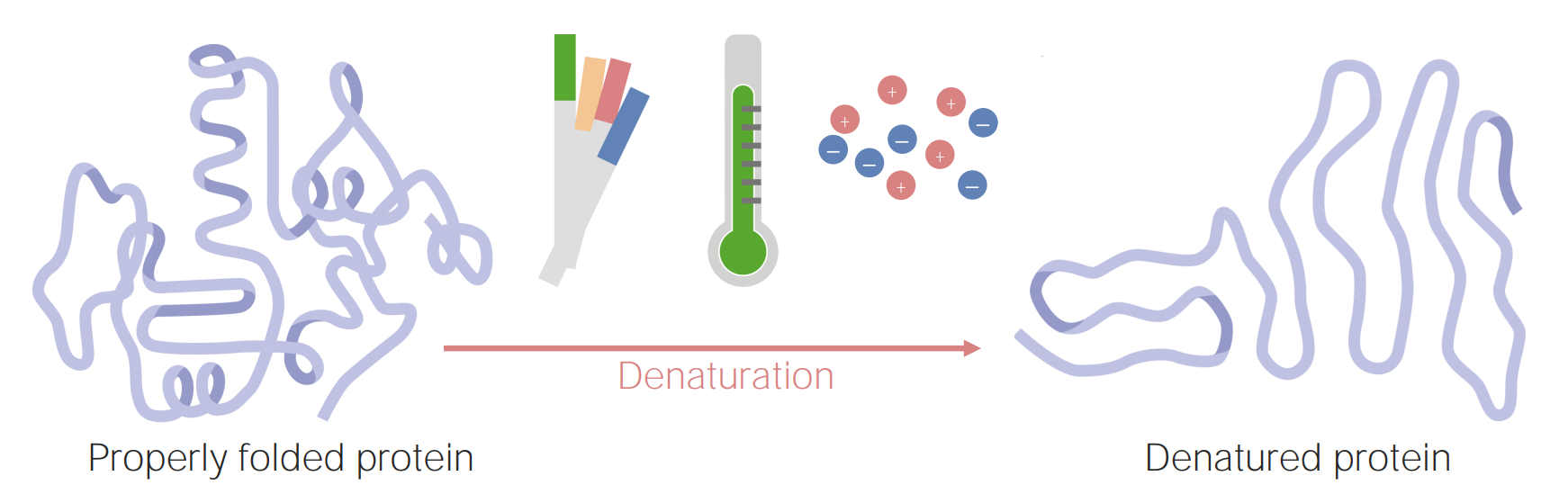
Las proteínas pueden desnaturalizarse (o desdoblarse) como resultado de cambios en el pH, la temperatura o la concentración iónica.
Imagen por Lecturio.La estructura única de una proteína (primaria, secundaria, terciaria y cuaternaria) le otorgará propiedades físicas y químicas que son importantes para la función de la proteína. Algunas de estas propiedades incluyen:
Las proteínas tienen una amplia gama de funciones en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum el cuerpo, que incluyen:
| Enzima | Zimógeno (precursor) | Activada por | Notas sobre la actividad |
|---|---|---|---|
| Enzimas gástricas secretadas hacia el estómago | |||
| Pepsina | Pepsinógeno | Ácido clorhídrico | Más eficiente entre los LOS Neisseria aminoácidos hidrófobos |
| Enzimas pancreáticas secretadas hacia el duodeno | |||
| Tripsina | Tripsinógeno | Enteropeptidasa | |
| Quimotripsina | Quimotripsinógeno | Tripsina | Más eficiente entre los LOS Neisseria aminoácidos hidrófobos |
| Carboxipeptidasa | Procarboxipeptidasa | Tripsina |
|
| Elastasa | Proelastasa | Tripsina | Igual que la carboxipeptidasa |
| Enzimas unidas al AL Amyloidosis borde en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum cepillo de los LOS Neisseria enterocitos en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum el intestino delgado | |||
| Aminopeptidasa | N/A | N/A | Descompone péptidos pequeños desde su extremo amino (i.e., N-terminal) |
| Dipeptidasa | N/A | N/A | Rompe enlaces peptídicos entre 2 aminoácidos → 2 aminoácidos individuales |
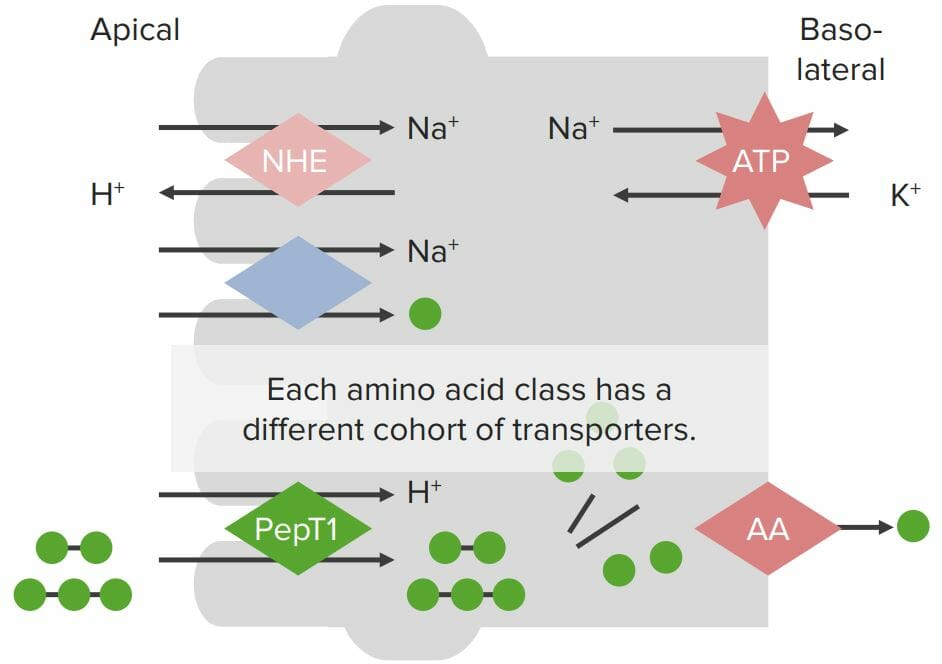
Proteínas de transporte en las membranas de los enterocitos involucradas en la absorción de proteínas:
La Na+/K+-ATPasa en la membrana basolateral genera un gradiente de Na+ dentro de la célula. Un intercambiador de Na+/H+ (NHE, por sus siglas en inglés) en la membrana apical también genera el gradiente de H+. Los aminoácidos individuales (AA; bolas verdes) se absorben a través de un cotransportador de Na+/AA, donde el Na+ fluye a través de la membrana apical hacia los enterocitos siguiendo su gradiente de concentración, trayendo consigo el AA (a pesar de moverse en contra del gradiente químico de los AA). Los péptidos pequeños se absorben a través del cotransportador H+/PepT con H+ fluyendo a favor de su gradiente de concentración hacia la célula, trayendo consigo los péptidos pequeños. Los péptidos se descomponen en AA individuales mediante peptidasas dentro de los enterocitos. Luego, todos los AA se absorben a través de transportadores especializados en la membrana basolateral.
El metabolismo de las proteínas se refiere a un grupo de procesos bioquímicos responsables tanto del anabolismo (síntesis de proteínas y aminoácidos) como del catabolismo (descomposición de proteínas y aminoácidos).
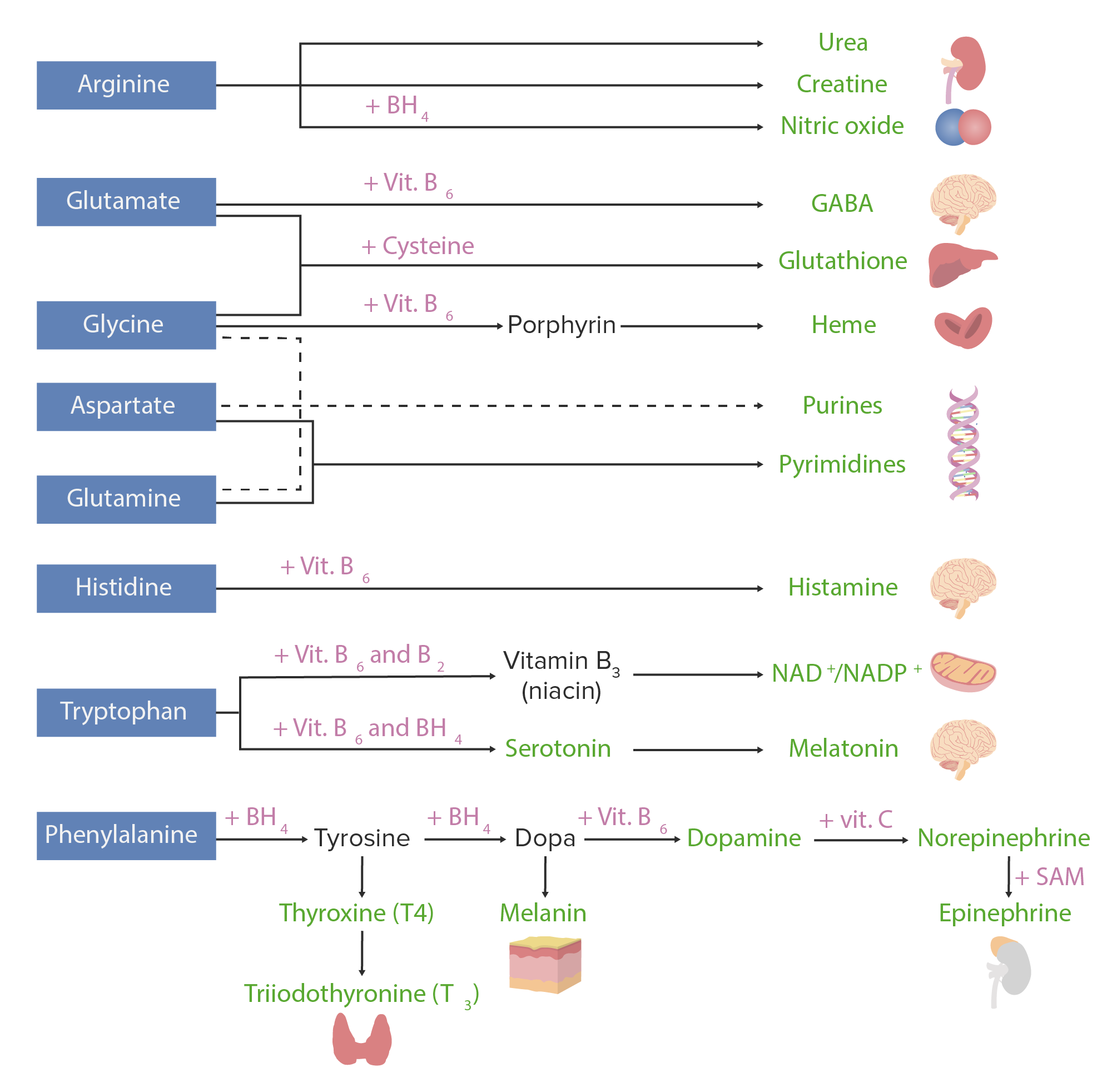
Derivados de los aminoácidos:
Los aminoácidos (en azul) se combinan con ciertos cofactores u otros sustratos (en rosa) para producir varias sustancias biológicamente importantes (en verde).
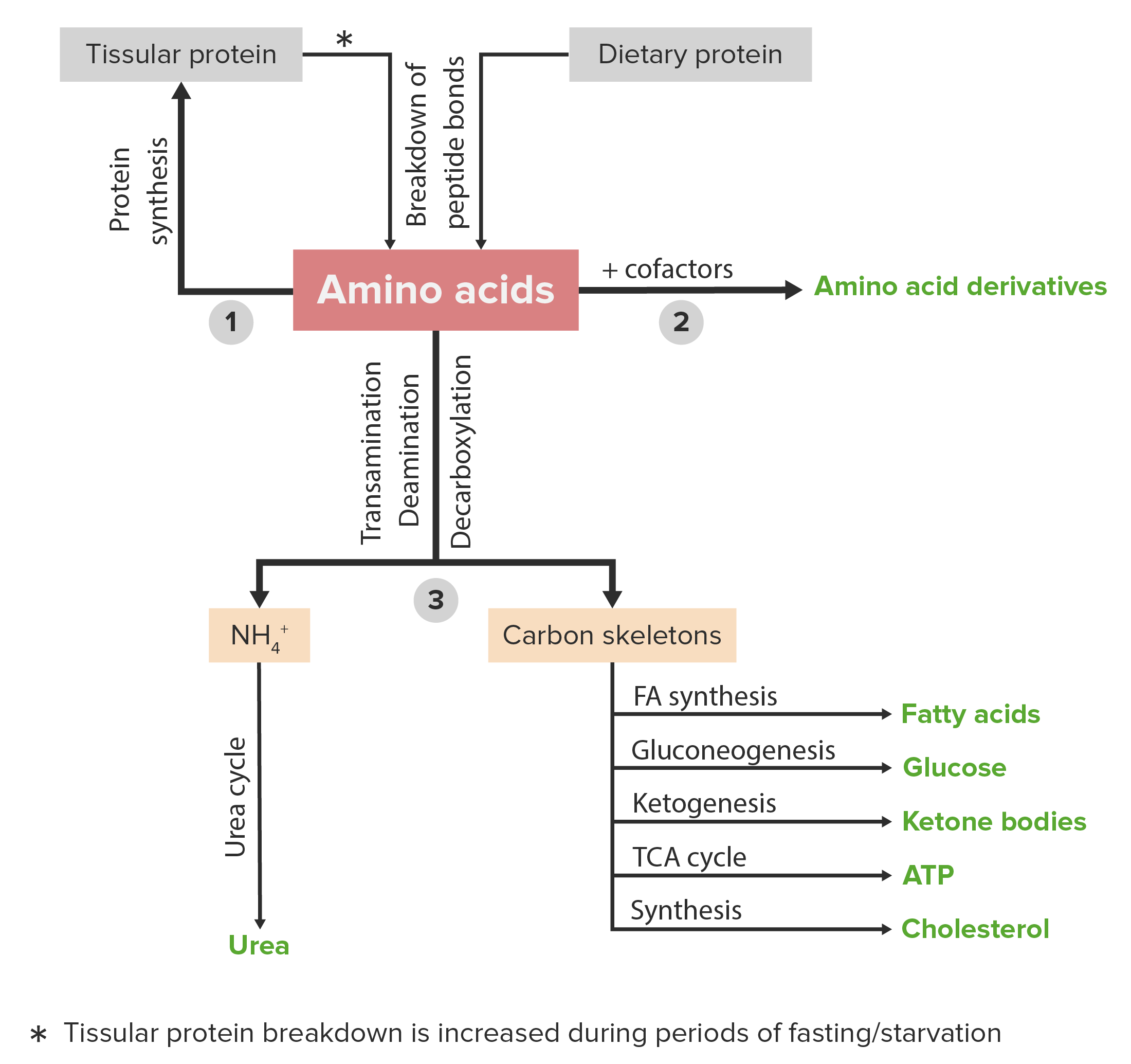
Diagrama esquemático del metabolismo de los aminoácidos, incluidas las 3 vías principales: reutilización en la síntesis de nuevas proteínas, unión con cofactores para producir derivados de aminoácidos y catabolismo. El catabolismo de los aminoácidos incluye la eliminación de grupos funcionales y la descomposición de los esqueletos de carbono.
Imagen por Lecturio.Un sinnúmero de trastornos clínicos son causados por anormalidades o deficiencias de proteínas y/o metabolismo proteico anormal. A continuación se enumeran algunos ejemplos.