


A púrpura trombocitopénica trombótica (PTT) é uma doença com risco de vida devido a uma deficiência congénita ou adquirida de ADAMTS-13, uma metaloproteinase que cliva multímeros do fator de von Willebrand (FVW). Os grandes multímeros agregam plaquetas excessivas resultando em trombose microvascular e num aumento do consumo de plaquetas. A apresentação clínica pode consistir em trombocitopenia, anemia Anemia Anemia is a condition in which individuals have low Hb levels, which can arise from various causes. Anemia is accompanied by a reduced number of RBCs and may manifest with fatigue, shortness of breath, pallor, and weakness. Subtypes are classified by the size of RBCs, chronicity, and etiology. Anemia: Overview and Types hemolítica, hematúria, sintomas gastrointestinais, sintomas neurológicos e envolvimento renal. O diagnóstico é estabelecido com base numa combinação de sintomas clínicos e exames laboratoriais. A púrpura trombocitopénica trombótica é uma emergência médica e quase sempre fatal se não se iniciar tratamento adequado atempadamente. O tratamento de emergência inclui plasmaférese e terapêutica imunossupressora.
Last updated: Dec 15, 2025
A púrpura trombocitopénica trombótica (PTT) é uma microangiopatia trombótica causada pela deficiência de ADAMTS-13, uma protease Protease Enzyme of the human immunodeficiency virus that is required for post-translational cleavage of gag and gag-pol precursor polyproteins into functional products needed for viral assembly. HIV protease is an aspartic protease encoded by the amino terminus of the pol gene. HIV Infection and AIDS de clivagem do fator de von Willebrand (FVW).
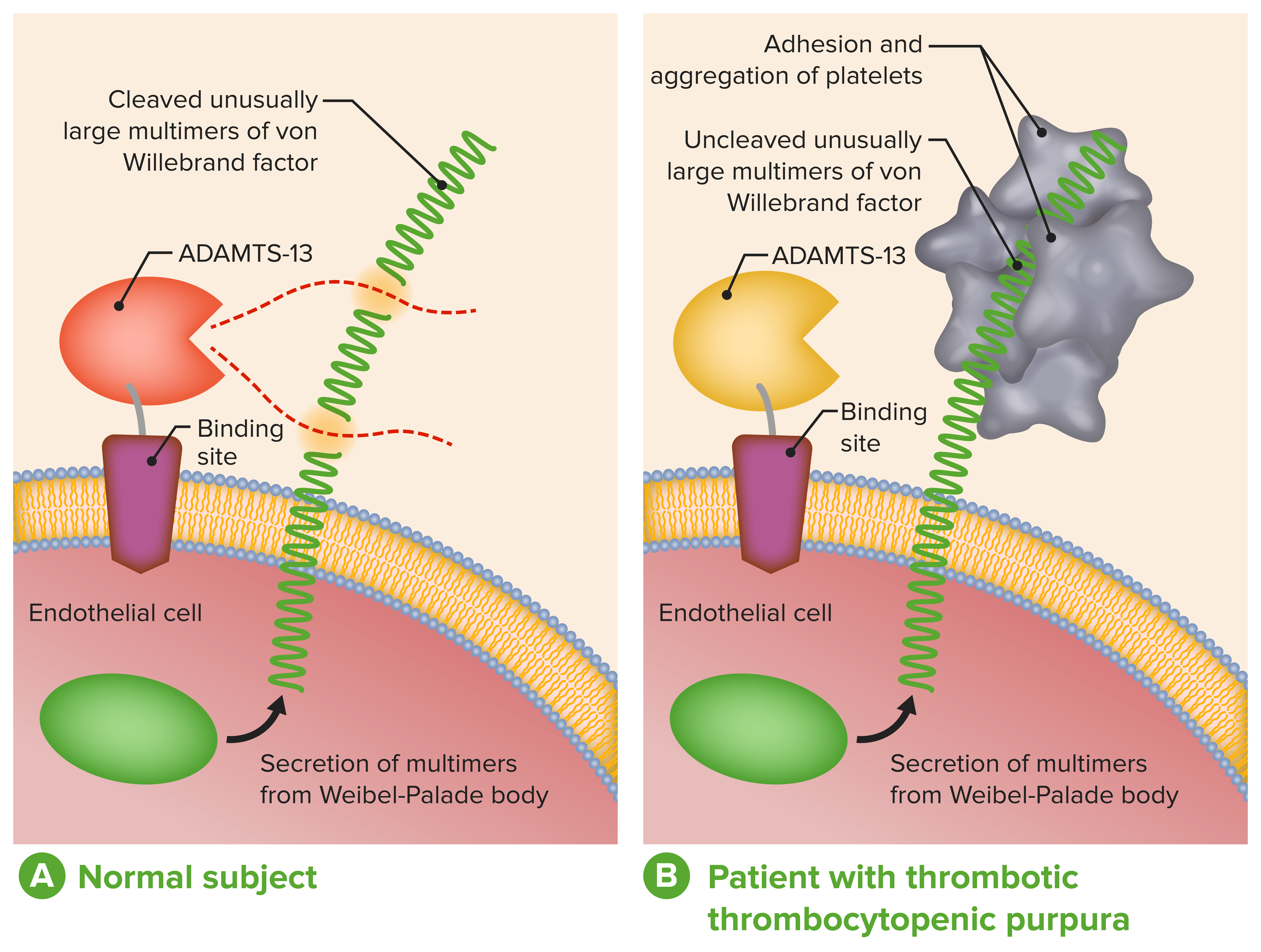
ADAMTS-13 no diagnóstico e tratamento de microangiopatias trombóticas:
A: Função fisiológica da ADAMTS-13 normal
B: Atividade da ADAMTS-13 ausente ou severamente reduzida em pacientes com PTT impede a clivagem oportuna de multímeros de FVW anormalmente grandes durante a secreção pelas células endoteliais.
Pêntade da PTT clássica:
Manifestações de órgãos-alvo:
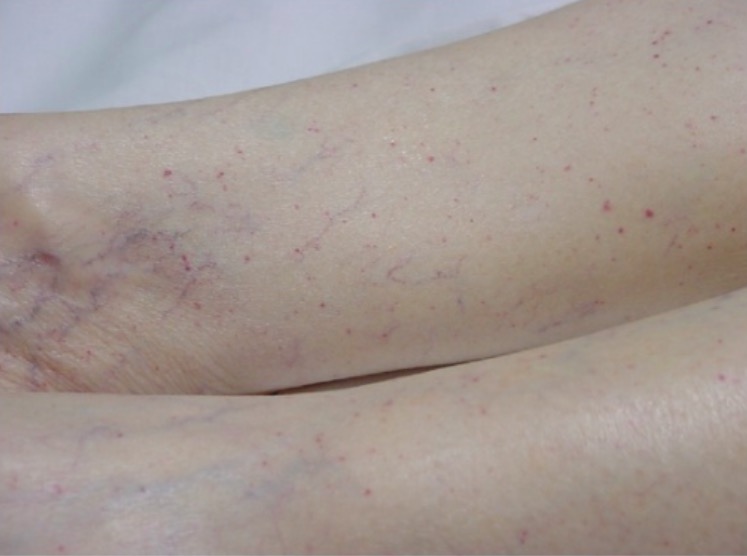
Erupção petequial típica nas extremidades inferiores de um paciente
Imagem : “Typical petechial rash” por Department of Emergency Medicine, University of Florida, Gainesville, FL 32610, USA. Licença: CC BY 3.0Ao contrário da PTT adquirida, a PTT congénita não apresenta episódios agudos.
Os sintomas são geralmente ligeiros, vagos e contínuos:
As exacerbações graves e com risco de vida são mais MAIS Androgen Insensitivity Syndrome prováveis durante 2 períodos da vida:
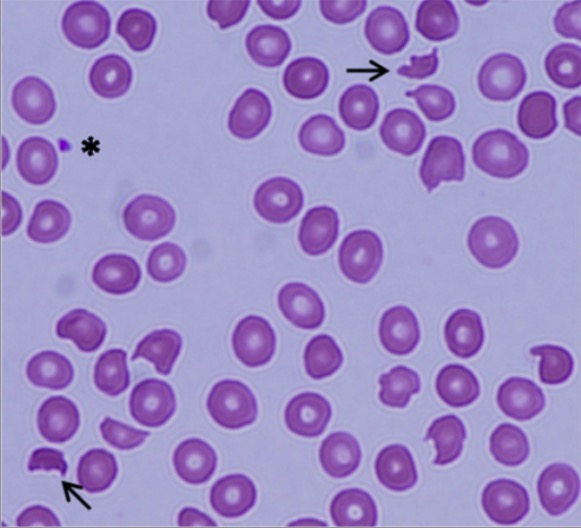
Púrpura trombocitopénica trombótica (PTT):
Esfregaço de sangue periférico mostrando esquizócitos (setas) e uma diminuição acentuada no número de plaquetas, indicando anemia hemolítica microangiopática
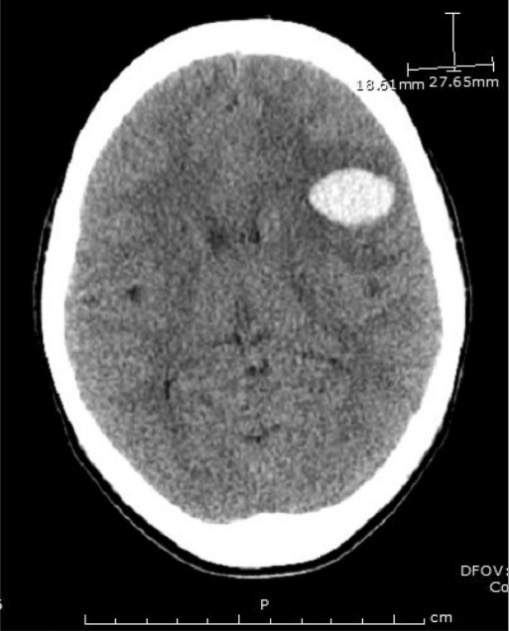
TC de crânio num paciente demonstrando a maior de 2 hemorragias frontoparietais
Imagem : “Emergent head CT demonstrating the larger of two frontoparietal hemorrhages” por Divisions of Infectious Disease, The George Washington University, School of Medicine and Health Sciences, Washington, DC, USA. Licença: CC BY 3.0O score PLASMIC prediz a probabilidade de PTT na presença de esquizócitos no esfregaço de sangue periférico.
Critérios PLASMIC (1 ponto por cada):
Pontuação:
