La inmunodeficiencia combinada grave, también llamada "enfermedad del niño burbuja", es un trastorno genético poco frecuente en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum el que el desarrollo de las células B y T funcionales se ve VE Ventilation: Mechanics of Breathing alterado debido a varias mutaciones genéticas que dan lugar a una función inmunitaria reducida o ausente. Es la forma más grave de inmunodeficiencia primaria y se caracteriza por la alteración de las respuestas inmunes humoral y celular. Múltiples mutaciones pueden dar lugar a tipos heterogéneos de inmunodeficiencia combinada grave. Los LOS Neisseria pacientes presentan infecciones graves y recurrentes en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum los LOS Neisseria primeros meses de vida. El tratamiento incluye inmunoglobulinas intravenosas y trasplante de médula ósea. Si no se trata, la inmunodeficiencia combinada grave suele ser mortal en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum el 1er año de vida.
Last updated: Dec 15, 2025
Se conoce que al AL Amyloidosis menos 12 genes Genes A category of nucleic acid sequences that function as units of heredity and which code for the basic instructions for the development, reproduction, and maintenance of organisms. DNA Types and Structure de estar mutados causan immunodeficiencia combinada grave, incluyendo los LOS Neisseria que codifican para lo siguiente:
El patrón de herencia depende del tipo de mutación genética:
| Fenotipo | Defecto genético |
|---|---|
| T–B+NK– | γC, JAK3 JAK3 A janus kinase subtype that is predominantly expressed in hematopoietic cells. It is involved in signaling from a broad variety of cytokine receptors including ones that utilize the interleukin receptor common gamma subunit. Severe Combined Immunodeficiency (SCID) |
| T–B–NK+ | RAG-1, RAG-2, Artemis, ADN ligasa IV, Cernunnos, ADN PKcs |
| T–B+NK+ | IL-7Rα, CD3δ, CD3ζ, Coronin-1A, ZAP-70, CD45 |
| T–B–NK– | ADA ADA An enzyme that catalyzes the hydrolysis of adenosine to inosine with the elimination of ammonia. Purine and Pyrimidine Metabolism, AK2 |
| Síndrome | Defecto |
|---|---|
| Inmunodeficiencia combinada grave ligada al AL Amyloidosis cromosoma X | Mutaciones en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum el gen que codifica la cadena γ común, una proteína que comparten los LOS Neisseria receptores de las interleucinas IL-2, IL-4, IL-7 IL-7 A proinflammatory cytokine produced primarily by T-lymphocytes or their precursors. Several subtypes of interleukin-17 have been identified, each of which is a product of a unique gene. Severe Combined Immunodeficiency (SCID), IL-9, IL-15 e IL-21 |
| Deficiencia de adenosina deaminasa |
|
| Deficiencia de purina nucleósido fosforilasa ( PNP PNP An enzyme that catalyzes the reaction between a purine nucleoside and orthophosphate to form a free purine plus ribose-5-phosphate. Purine and Pyrimidine Metabolism) | Trastorno autosómico recesivo que implica mutaciones del gen PNP PNP An enzyme that catalyzes the reaction between a purine nucleoside and orthophosphate to form a free purine plus ribose-5-phosphate. Purine and Pyrimidine Metabolism |
| Disgenesia reticular | Incapacidad de los LOS Neisseria precursores de granulocitos para formar gránulos, secundaria a un mal funcionamiento de la adenilato quinasa 2 mitocondrial |
| Síndrome de Omenn |
|
| Síndrome de los LOS Neisseria linfocitos desnudos |
|
| JAK3 JAK3 A janus kinase subtype that is predominantly expressed in hematopoietic cells. It is involved in signaling from a broad variety of cytokine receptors including ones that utilize the interleukin receptor common gamma subunit. Severe Combined Immunodeficiency (SCID) | JAK3 JAK3 A janus kinase subtype that is predominantly expressed in hematopoietic cells. It is involved in signaling from a broad variety of cytokine receptors including ones that utilize the interleukin receptor common gamma subunit. Severe Combined Immunodeficiency (SCID) es una enzima que media la transducción de las señales γc. |
La enfermedad suele presentarse en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum la primera infancia (2–6 meses). Se observan infecciones oportunistas graves y recurrentes en EN Erythema nodosum is an immune-mediated panniculitis (inflammation of the subcutaneous fat) caused by a type IV (delayed-type) hypersensitivity reaction. It commonly manifests in young women as tender, erythematous nodules on the shins. Erythema Nodosum los LOS Neisseria individuos afectados.
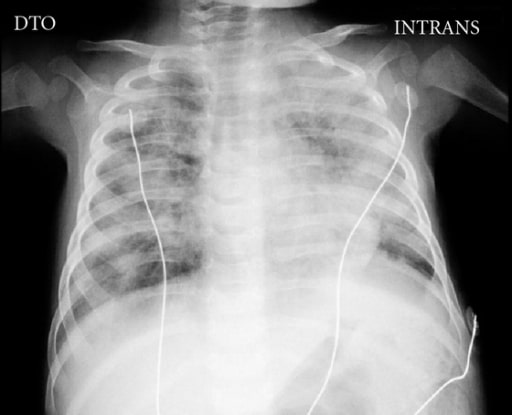
Radiografía de tórax de un niño de 5 meses con inmunodeficiencia combinada grave complicada por la enfermedad diseminada del bacilo de Calmette-Guérin que muestra la ausencia de timo y áreas bilaterales de opacidades
Imagen: “Chest X-ray” por Pediatric Intensive Care Unit, Hospital Dona Estefânia, 1169-045 Lisboa, Portugal. License: CC BY 3.0
Históricamente se utilizaron medidas extremas para lograr un aislamiento eficaz de los pacientes con inmunodeficiencia combinada grave, lo que le concedió el nombre de “enfermedad del niño burbuja”.
Imagen: “David Vetter y John Montgomery” por Jeremy112233. Licencia: Dominio PúblicoLas siguientes condiciones son diagnósticos diferenciales para la inmunodeficiencia combinada grave: